






最新资讯



-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
NRF2和KEAP1在慢性疾病中的治疗靶向性
发表于:2019-04-02 作者:admin 来源:本站 点击量:13252
Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov. 2019 Jan 4. doi: 10.1038/s41573-018-0008-x. [Epub ahead of print] Review. PubMed PMID: 30610225.
摘要:转录因子NF-E2 p45相关因子2(NRF2;由NFE2L2编码)及其主要负调节因子,E3连接酶受体Kelch样ECH相关蛋白1(KEAP1)是维持氧化还原、代谢和蛋白质稳态以及炎症调节的关键。因此,激活NRF2可对多种疾病起到细胞保护作用,包括肺和肝的慢性疾病、自身免疫病、神经退行性疾病和代谢性疾病以及癌症启动。一种NRF2激活剂已获得临床批准,并且基于半胱氨酸传感器KEAP1的几种亲电修饰剂及其与NRF2相互作用的抑制剂目前正在临床开发中。然而,在靶向特异性、药效学特性、疗效和安全性方面仍存在挑战。
前言
转录因子NF-E2 p45相关因子2(NRF2;由NFE2L2编码)于1994年作为人类cap 'n' collar(CNC)碱性区域亮氨酸拉链转录因子家族成员被发现(1)。随后的研究,包括NRF2敲除小鼠的产生,确定NRF2调控约250个基因的表达,调控的基因在其启动子调节区中含有增强子序列,称为抗氧化应答元件(ARE)(2, 3)。这些基因编码一个合作酶网络,参与内生和外生生物转化反应、抗氧化代谢、碳水化合物和脂类的中间代谢、铁的分解代谢、蛋白质降解和炎症调节(4)。通过这个转录网络,NRF2能够协调多方面应对各种形式的应激,维持稳定的内部环境。
1999年,Kelch样ECH相关蛋白1(KEAP1)被鉴定为NRF2的抑制因子(5),又过了5年才被报道为E3泛素连接酶底物适配器,靶向NRF2进行蛋白酶体的快速降解(6-8)。因此,KEAP1确保在无应激条件下,NRF2是低丰度的蛋白质,具有仅约15-40分钟的有限半衰期,这取决于细胞类型。然而,重要的是,KEAP1含有几个高活性的半胱氨酸,经亲电子分子修饰后,可防止其靶向NRF2进行蛋白酶体降解,从而导致NRF2蛋白在氧化还原应激下快速核聚集,并在与小肌肉筋膜纤维肉瘤癌基因同源蛋白(sMAF)二聚化后,通过NRF2-sMAF诱导含有ARE的基因(9,10)。
现在人们普遍认识到,由于NRF2-靶基因编码的蛋白质的多功能和全面的细胞保护作用(包括抗氧化、解毒和抗炎),因此,NRF2-KEAP1信号通路对于预防以氧化应激和炎症为基础病理特征的多种疾病至关重要。这些疾病包括代谢、炎症和自身免疫性疾病,肺、肝、肾、胃肠道和心血管系统疾病以及神经疾病(9, 11)。NRF2在这些非肿瘤性疾病中的保护作用的实验证据将在本综述的后面讨论。我们还讨论了NRF2在癌症中的作用,这是一个需要深入研究的问题(12)。
通过单粒子电子显微镜获得的KEAP1二聚体3D重建显示,该蛋白具有叉状茎结构,包括两个大球体,它们包围了中间区域(IVR)、Kelch和羧基末端结构域,而二聚化由BTB域介导(图1a)(13)。考虑到KEAP1包含许多不同且自主运作的半胱氨酸传感器(C151、C226、C273、C288和C613)(14-16),现有证据表明,NRF2和KEAP1整合了一氧化氮、Zn2+、烯醛信号传导与氧化还原信号传导一起提供了一种可能至少部分进化的机制,以允许快速适应饮食中遇到的植物化学物质。许多植物来源的亲电异种生物激活NRF2-KEAP1信号通路以引发细胞保护反应,KEAP1中的高活性半胱氨酸起直接传感器的作用(17-19)。这些传感器与亲电子试剂的高反应性使得KEAP1在没有非特异性硫醇修饰的情况下进行靶向治疗成为可能(图1b)。因此,已经描述了许多天然存在的NRF2靶基因诱导剂(20),并且新的诱导剂在不断被发现。
尽管大量学术实验室参与了NRF2激活剂的鉴定,但从历史上看,制药公司在这个令人兴奋和充满希望领域的参与度却相当低。业界之所以不愿将NRF2-keap1信号通路视为有价值的药物靶点,部分原因可能是其广泛的生物化学活性,而不仅仅是控制由NRF2直接或间接调控的抗氧化系统。NRF2激活剂的各种直接和间接效应有时被解释为非特异性效应的证据。然而,KEAP1中几种不同的基于硫醇的氧化还原开关的进化允许哺乳动物适应广泛的植物化学物质,这为改变NRF2的激活引起某些“脱靶”效应并采用更广泛视野的观念提供了理论基础。NRF2的激活产生有益的“多靶点”细胞保护作用(11)。尤其是NRF2激活的多靶点效应包括维持氧化还原信号、增强异种生物转化、控制和解决炎症、抑制糖异生和肝脂肪生成、支持蛋白稳定和抑制纤维化(9)。在这方面,值得注意的是,大多数慢性疾病,特别是老年人的慢性病,没有独特的病因学,也没有单一的病理表型,因此,它们可能通过激活NRF2的药物得到最有效的治疗。事实上,与更有针对性的治疗干预相比,NRF2激活后广泛的细胞保护机制的刺激不太可能被致病过程有效地淹没或规避。
这种对NRF2作为潜在药物靶标的广泛认可引起了制药业的兴趣,并导致对NRF2调节剂临床开发的大量投资(21)。NRF2激活剂富马酸二甲酯(DMF;目前,商标名如Fumaderm或Tecfidera)已被临床用于治疗银屑病和复发性多发性硬化症(MS)(表1),其目前的分布情况仍是其他制药公司希望达到的目标。目前,多种不同化学类型的NRF2激活剂处于临床开发的不同阶段,如靶向KEAP1半胱氨酸的富马酸衍生物、异硫氰酸硫萝卜硫素(SFN;4‐异硫氰酸甲酰丁酯)、氰烯酮三萜、硝基脂肪酸和羟胺。其他候选药物包括破坏NRF2-KEAP1蛋白-蛋白相互作用(PPIs)的非亲电化合物,以及具有keap1独立作用模式的分子。NRF2的抑制剂也在研究中,但尚未进行临床试验。本综述概述了这些候选药物,并强调了针对NRF2-KEAP1信号通路不寻常的性质和至关重要的意义。本文描述了NRF2在细胞氧化还原、代谢和蛋白稳态以及炎症调控中的生理作用,并为NRF2在非肿瘤性疾病中的保护作用提供了实验证据。评价了目前临床开发的各种小分子NRF2活性药理调节剂的优缺点。最后,强调了与NRF2调节剂开发相关的持续挑战,包括靶点特异性、药效学评估、生物利用度、有效性和安全性。
NRF2-KEAP1信号通路的生理作用
氧化还原、代谢和蛋白质稳态
自从其最初被发现参与生物转化反应,调节某些细胞色素P450氧化还原酶、结合酶和ABC转运蛋白的表达(表2),NRF2参与控制细胞内氧化还原环境的作用变得越来越明显,最终将NRF2-KEAP1信号通路识别为“硫醇驱动的总开关”用于“整个系统范围的氧化应激反应”(22)。NRF2调控参与NADPH生成的4个基因(葡萄糖6-磷酸脱氢酶、6-磷酸葡萄糖酸脱氢酶、苹果酸酶1和异柠檬酸脱氢酶1)的表达(23–26)。这种还原当量的供应进一步被大量氧化还原反应所利用,其中许多氧化还原反应也受到NRF2的调节。因此,NRF2调控氧化还原缓冲物还原型谷胱甘肽(GSH;其氧化形式为GSSG),如谷氨酸半胱氨酸连接酶、谷胱甘肽还原酶、谷胱甘肽过氧化物酶和数个谷胱甘肽s -转移酶的催化和调节亚基(4)。此外,氧化还原蛋白家族中的许多辅助蛋白,如硫氧还蛋白、硫氧还蛋白还原酶、过氧化物还蛋白和硫还蛋白,都受到NRF2的调控,提供区域活性氧生成的区域化传感和信号转导(ROS;即过氧化氢(H2O2)羟基和超氧自由基 )(27-29)。除了这些对氧化还原状态的直接影响外,NRF2还调节编码酶的基因,这些酶可阻止醌参与氧化还原循环反应和谷胱甘肽耗竭(通过NAD(P)H:醌氧化还原酶1(NQO1))或参与间接生产胆红素(血红素加氧酶1(HMOX1)和胆绿素还原酶(BVR)),是最有效的非极性生理抗氧化剂(30)。最后,NRF2通过与戊糖磷酸途径和糖酵解的相互作用,影响中间代谢,增加底物的可用性,减少线粒体呼吸链的等效物(31),并维持线粒体DNA (mtDNA)的完整性(32)。
NRF2在氧化还原改变或营养缺乏期间参与清除氧化或其他损伤的蛋白质和细胞器。因此,自噬转运蛋白泛素结合蛋白p62(由SQSTM1编码)是NRF2的转录靶点,它通过其泛素关联域(UBA)与泛素化产物相互作用,并通过其LC3相互作用的基序将其招募到自噬体中(33, 34)。此外,KEAP1与p62结合(33, 35),最近的一项研究发现KEAP1-CULLIN 3(CUL3)在其UBA结构域中的K420位点泛素化p62,从而促进p62的螯合活性(36)。在其一级结构中,p62含有ETGE样基序STGE,在被mTORC1磷酸化后产生识别位点与KEAP1对接(37)。然后,KEAP1被转运到自噬体降解,从而使NRF2积累(38)。此外,一些自噬基因似乎在其启动子区域含有序列,有报道称NRF2同时激活伴侣介导的自噬(39)和巨自噬(40,41)。NRF2在氧化条件下在蛋白质稳态中的其他作用通过未折叠蛋白质反应的激活(42,43)及其调节蛋白酶体亚基表达的能力来证明(44,45)。
至少在脑和血液中,编码NRF2的mRNA丰度高于KEAP1的mRNA(图2);相反,KEAP1蛋白的半衰期比NRF2的半衰期长(表2)。这些观察结果强调了两种蛋白质的不同转换率——KEAP1缓慢,NRF2快。此外,尽管NRF2和KEAP1普遍表达,但特定细胞类型主要负责稳态适应并呈现不同的表达水平(图2)。因此,单核细胞和嗜中性粒细胞在血细胞中表现出最高水平的NRF2,表明先天免疫系统的免疫调节作用(见下文)。在大脑中,星形胶质细胞和神经元之间的相互作用将中间代谢与氧化还原稳态结合起来,至少部分通过NRF2实现(46)。此外,神经元中的谷氨酸能神经传递导致产生由神经元GSH池控制的ROS。当该池浓度较低时,邻近的星形胶质细胞通过向神经元提供GSH前体甘氨酸、谷氨酸/谷氨酰胺和半胱氨酸来促进其恢复(47)。毫不奇怪,星形胶质细胞中NRF2水平很高(图2)。此外,与神经元相比,小胶质细胞作为单核细胞谱系的一部分表达高水平的NRF2。
控制炎症
NRF2在单核细胞和粒细胞中含量丰富(图2),提示NRF2参与了这些细胞类型驱动的免疫应答。敲除NRF2的小鼠对败血症休克高度敏感(48),并且在创伤愈合过程中表现为持续性炎症(49, 50)。而NRF2的基因或药理激活抑制了促炎细胞因子的产生(51 - 56)。在人类细胞中,KEAP1的破坏通过减弱髓系白细胞(57)和巨噬细胞(58)的炎症反应来预防败血症。特别有趣的是,NFE2L2中的一个多态性与转录活性降低相关,与炎症性肠病(IBD)(59)和慢性胃炎(60)风险增加相关。如下所示,NRF2的强抗炎活性至少可归因于三个独立的机制:氧化还原代谢的调节;与核因子-κB(NF-κB)的相互作用;对促炎基因的直接调控。
炎症与局部和全身病理水平的ROS积累增加有关,这可能损害氧化还原信号(61)。线粒体功能障碍和NADPH氧化酶不受控制的激活是炎症细胞中ROS生成升高的主要因素,线粒体ROS引起mtDNA损伤和释放,从而形成恶性循环,进一步ROS的产生和炎性小体的激活,最终导致器官功能衰竭(62)。NRF2的氧化还原调节活性是阻止炎症恶化和随后组织损伤的恶性循环中的重要突破(63)。
NF-κB和NRF2相互作用。几个NF-κB功能结合位点已确定在NFE2L2基因启动子区(64)。另一方面,NRF2抑制NF-κB转录活性。用脂多糖(LPS)或TNF处理NRF2缺陷小鼠增加了NF-κB转录的表达,表明NRF2对NF-κB的抑制作用(48)。该发现的可能解释是NF-κB抑制剂(IκB)在NRF2缺陷细胞中高度磷酸化并且经历快速蛋白酶体降解,从而减少对NF-κB的抑制(48)。此外,LPS显示同时激活NF-κB介导的快速反应和NRF2介导的缓慢反应,最初的促炎反应由NF-κB驱动,然后当NRF2活性最大时随后被抑制(65)。NRF2-KEAP1信号通路抑制NF-κB信号传导的其他机制包括NRF2和p65之间与p300竞争性结合(66)和KEAP1对IκB激酶-β(IKKβ)的降解(67, 68)。
几种巨噬细胞特异性基因含有ARE序列,因此受NRF2调节。这些包括MARCO(细菌吞噬作用所需的受体)和CD36(氧化低密度脂蛋白的清道夫受体)(69, 70)。 另一方面,NRF2以不依赖ARE的方式结合编码IL-6和IL-1β促炎基因调节区,并阻止RNA聚合酶II开始转录募集(52)。
最近的研究表明巨噬细胞代谢与NRF2引起的抗炎反应有关。线粒体代谢物亚甲基丁二酸是由三羧酸循环在促炎刺激下产生的,在巨噬细胞活化过程中会显著增加(71, 72)。反过来,亚甲基丁二酸烷基化几个KEAP1半胱氨酸传感器,包括关键C151、C273和C288残基,从而激活NRF2并抑制促炎细胞因子如IL-6和IL-1β的转录(71)。因此,亚甲基丁二酸是KEAP1的内源性调节剂,在炎症的控制过程中起到“关闭开关”的作用。此外,通过细胞可渗透的亚甲基丁二酸衍生物4-octylitaconate或SFN激活NRF2,通过降低mRNA稳定性来抑制干扰素基因(STING)的衔接子分子刺激物的表达,从而抑制I型干扰素(IFN)的产生(73,74)。
NRF2-KEAP1信号通路和非肿瘤性疾病
在细胞和动物模型中已经发现了大量关于NRF2对多种慢性疾病保护作用的信息,许多研究使用了NRF2敲除小鼠(75)。关于人类,最近的一项研究为使用系统医学方法分析NRF2对抗慢性病奠定了基础,并且基于NRF2与其他分子(NRF2相互作用组)的连通性以及经验证据,建立了第一个NRF2相关疾病(NRF2病变)的图谱(9)。NFE2L2的功能性遗传变异与疾病风险之间的关联研究不断提供有关NRF2在确定人类对慢性病易感性中作用的令人信服的证据。此外,这些遗传学研究支持这样一种假设,即对于NFE2L2的某些功能变化,模仿NRF2表达的微小变化的药物可能具有治疗价值。下面,我们将简要描述NRF2参与人类慢性疾病的一些里程碑性的成果,并重点介绍现有的遗传学证据。
自身免疫性疾病
氧化组织损伤和细胞凋亡导致半抗原或受损大分子的形成,增加了自身免疫反应的风险。由于NRF2调节的酶在许多化学物质的解毒中起着关键作用,因此它提供了一种保护机制,防止对自身免疫发病机制的环境易感性(79)。此外,NRF2抑制促炎T辅助细胞1(TH1)和TH17细胞反应并激活免疫抑制调节性T细胞(Treg)和TH2细胞(80)。NRF2在自身免疫中的相关性已经在实验性自身免疫性脑脊髓炎(EAE)中进行了广泛研究,其是MS的小鼠模型,MS是一种慢性炎性疾病,其特征在于自身反应性免疫细胞浸润到中枢神经系统中。在EAE小鼠模型中,NRF2的缺乏加剧了疾病(81),而KEAP1的敲低(52)或用一系列NRF2激活剂(包括氰基三萜类化合物和SFN)(82,83)治疗减弱了其严重性。在人类中,通过IFNβ治疗患者基因表达谱确定NRF2是长期抗氧化反应和神经元保护的潜在介质(84)。如下所述,一些生物制药公司正在利用强有力的证据表明NRF2的活化在MS中具有治疗价值。事实上,迄今为止唯一获得美国食品药品管理局(FDA)和欧洲药品管理局(EMA)DMF批准的NRF2激活剂正在由Biogen销售,用于治疗缓解复发型MS和银屑病(85)。NRF2的保护作用已被建议用于其他自身免疫性疾病,如系统性红斑狼疮(86, 87)、干燥综合征(86)、类风湿性关节炎(88, 89)、白癜风(90-92)和STING依赖性干扰素病(74)。
呼吸系统疾病
肺通过释放促炎细胞因子和趋化因子对包括烟雾在内的各种环境毒物作出反应。这一主要效应通过循环单核细胞、中性粒细胞和T细胞的募集产生渐进的低度炎症反应,这些细胞释放额外的炎症介质、ROS和金属蛋白酶。最终结果是肺实质受损的恶性循环,导致慢性阻塞性肺病(COPD)或肺气肿(93)。NRF2减轻由ROS和炎症引起的肺实质负担。香烟烟雾是COPD和肺气肿发展的主要危险因素。
NFE2L2-/-小鼠对香烟烟雾引起的肺气肿的发展比野生型更敏感(94)。此外,长期暴露于香烟烟雾后肺气肿的发展与细胞保护基因的表达下降有关(95),而五环氰基酮CDDO-咪唑啉保护NFE2L2+/+小鼠对抗香烟烟雾诱导的氧化应激、肺泡细胞凋亡、肺泡破坏和肺动脉高压,而不是NFE2L2-/-小鼠(96)。NRF2的遗传上调(通过Clara细胞中的Keap1缺失,其在鼠上呼吸道中是丰富的)具有类似的保护作用(97)。
在人类中,与没有肺气肿的患者相比,吸烟相关肺气肿患者的肺泡巨噬细胞中NRF2转录信号降低(98)。人类NFE2L2基因启动子包含一个含有三个单核苷酸多态性(SNP)序列的单倍型和一个调控NRF2表达的三重重复多态性。具有低至中等启动子活性的COPD患者更容易发生呼吸衰竭(99)。SFN对NRF2的药理学激活,或一种干扰KEAP1与NRF2相互作用的化合物,逆转培养肺泡巨噬细胞和从COPD患者中分离的单核细胞来源巨噬细胞受损的细菌吞噬作用(70,100)。然而,每日口服一定剂量SFN(西兰花芽苗提取物)的慢性阻塞性肺病患者并没有导致NRF2依赖性基因表达或肺泡巨噬细胞和支气管上皮细胞炎症标志物的一致性变化(101),说明将细胞培养研究结果转化为人类结果的一些挑战。其他慢性肺病,如特发性肺纤维化、慢性结节病和过敏性肺炎,被发现在支气管肺泡灌洗液中表现出NRF2表达增加和内源性抗氧化剂水平增加,表明对病理性ROS水平的适应性反应不成功(102)。
胃肠疾病
胃肠道经常受到外来生物的挑战,这可能导致形成病理水平的ROS,并可能引发IBD(103)。胃肠道也是膳食及其合成类似物在药理学上激活NRF2的重要部位之一(23, 104-106)。在小鼠中,右旋糖酐硫酸钠(DSS)诱导的实验性结肠炎在NRF2缺陷小鼠中引起的IBD要比野生型小鼠严重(107)。在人类中,NRF2在肠细胞对炎症应激适应的相关性在IBD患者的一项全面转录组研究中得到证实,证明了NRF2在减弱肠道巨噬细胞应激反应中的作用(108)。重要的是,在一组日本患者中发现了NFE2L2基因中特定SNP与溃疡性结肠炎发病风险之间的遗传关联(109)。NFE2L2启动子中3个SNP功能单倍体NRF2表达略有降低,与胃黏膜炎症和幽门螺杆菌感染相关消化性溃疡的发生发展有关(60)。
肝脏是抵御食物、外来生物制剂和口服活性药物的第一道防线,因为从消化道流出的大部分血液通过肝门静脉直接流到肝脏。早期使用NFE2L2-/-小鼠证实了NRF2对对乙酰氨基酚的保肝作用(110),NRF2的遗传活化也是如此(111)。正如可能预期的那样,喂食酒精(4-7天)的NFE2L2-/-小鼠比同样喂养的野生型小鼠明显更容易脂肪肝加重、肝脏炎症反应和肝功能衰竭,这在一定程度上可能是SREBP脂源性转录因子的激活和对乙醛解毒的相对无能(112)。通过观察NRF2在小鼠肝部分切除后对肝脏再生的阻碍,发现NRF2调控NOTCH1基因表达(113),这一机制也参与了NRF2介导的造血干细胞祖细胞功能改善和电离辐射后骨髓抑制(114)。在遗传性血色素沉着症伴铁超载的小鼠模型中(由于稳态铁调节基因突变),敲除NRF2增加了小鼠肝脏内导致肝纤维化的坏死炎症病变(115)。与这些观察结果一致,原发性胆管炎和肝硬化患者NRF2表达减少(116。)
代谢性疾病
代谢性疾病又称代谢综合征,以腹部肥胖、高血压、高甘油三酯血症、低水平高密度脂蛋白和空腹高血糖为特征。它与2型糖尿病(T2DM)和相关的合并症如血管功能障碍、心血管疾病、肾小球肾炎、非酒精性脂肪性肝炎(NASH;见下文)和认知障碍密切相关。尽管T2DM及其合并症的特征在于线粒体产生过量超氧化物(117),但NRF2缺失小鼠不会自发地发展成糖尿病。实际上,敲除小鼠表现出增加的胰岛素敏感性,这归因于ROS介导的蛋白酪氨酸激酶磷酸酶1B的抑制,该蛋白拮抗胰岛素信号(118)。相比之下,db/db背景下小鼠中KEAP1敲除或敲低对NRF2的遗传激活,或db/db小鼠中NRF2的药理活化,抑制了T2DM的发生(119)。因此,似乎当胰腺β细胞代谢挑战合成胰岛素以应对高血糖时,所产生的ROS负担可能会导致氧化损伤、损害反应、导致T2DM(120,121)。如果这个结论是正确的,那么很可能在饮食诱导的T2DM小鼠中,TBE-31或SFN激活NRF2后,胰腺功能的改善对葡萄糖处理的改善有显著的作用(122,123)以及富含SFN西兰花芽提取物对T2DM失调肥胖患者空腹血糖和糖化血红蛋白(HbA1c)的降低作用(123)。除了增加对胰腺β细胞的抗氧化应激能力之外,NRF2活化可通过增加糖原分支酶和磷酸化酶b激酶α的表达来抑制骨骼肌中的糖原分解,从而有助于改善全身葡萄糖稳态(119)。在糖尿病肾病小鼠模型中,SFN或肉桂醛对NRF2的药理激活已被证明可抑制氧化应激以及转化生长因子β1信号传导和纤维化(124)。
NRF2可能在胰岛素抵抗中发挥关键作用,因为它在糖尿病患者的单核细胞中表达很低(125)。令人信服的证据来自遗传关联研究,表明导致低NRF2表达的一些NFE2L2多态性导致病理性ROS和T2DM的风险增加(126-128)。关于并发症,由于KEAP1启动子中CpG岛的去甲基化导致的抗氧化保护失效与糖尿病白内障有关(129)。
NRF2也可能对NASH的发展具有保护作用。在使用缺乏蛋氨酸和胆碱饮食的NASH小鼠模型中,NRF2缺失小鼠比野生型小鼠死于该疾病的速度要快得多(130,131)。在更为生理的NASH模型中,喂食高脂肪饮食的NRF2缺失小鼠也会出现肝脏脂肪变性和炎症恶化,这表明需要NRF2来抑制这些症状的发生(118, 132)。此外,野生型而非NRF2敲除小鼠,在长期高脂肪、高果糖饮食使其变得肥胖和胰岛素抵抗后,通过三环氰基酮TBE-31对NRF2药理激活后,逆转了胰岛素抵抗,抑制肝脏脂肪变性,改善NASH和肝纤维化(122)。TBE-31对NRF2的药理激活拮抗SREBP1c和ChREBP及其脂质生物合成靶基因的表达,这可能涉及AMPK的激活(122, 133)。此外,在小鼠肝脏中,NRF2的基因激活可拮抗糖异生酶PEPCK和G6PC的表达,这可能也涉及AMPK(133)。在NASH患者的肝脏活检标本中,发现病理性ROS水平升高、NRF2基因表达增加,提示其试图减轻氧化和炎症(134)。
心血管疾病
ROS的产生和处理之间的不平衡导致破坏性氧化物质的过度形成,已被认为在许多心血管疾病中发挥作用,如高血压、动脉粥样硬化、糖尿病血管疾病、心肌缺血再灌注损伤和心力衰竭(135)。此外,血管壁内的低度持续性炎症是动脉粥样硬化易损斑块的形成过程和发展特点,所以动脉粥样硬化斑块易破裂,进而导致血栓形成和终末器官缺血。鉴于NRF2是抗氧化防御的关键调控因子,且具有直接的抗炎特性,因此可以合理地假设NRF2可以预防与ROS相关的心血管疾病。事实上,包括富马酸盐和硫化氢在内的NRF2激活剂在许多心血管疾病动物模型中显示出了保护作用,如心脏缺血再灌注损伤(136, 137)和心力衰竭(138, 139)。NRF2在高胆固醇血症小鼠模型动脉粥样硬化中的作用就不那么直接了:在骨髓来源细胞中NRF2的缺失加重了动脉粥样硬化(140, 141),而根据病变发展程度评估的NRF2全面缺失减轻了动脉粥样硬化(142-145),但增加了斑块炎症和易损性(145)。然而,一些NRF2激活剂已被证明在多种动脉粥样硬化动物模型中具有动脉粥样硬化保护作用(146-148)。由收缩性、细胞骨架蛋白和分子伴侣蛋白突变引起的心脏疾病约占遗传性心肌疾病的30%(149)。在某些情况下,还原而不是氧化应激,再加上蛋白质聚集,与心肌病有关。血液透析患者全身炎症和病理ROS的形成与NRF2的下调有关(150),位于NFE2L2启动子的多态性与这些患者死亡率的增加有关(151)。NRF2信号通路的破坏可防止还原性应激并延缓过量表达心脏中CRYAB(α-结晶B链)小鼠蛋白质性心脏病(152)。因此,正如这篇综述所强调的,针对NRF2的环境非常重要——越多不一定越好。
神经退行性疾病
在各种形式的神经退行性疾病中,NRF2和蛋白稳定之间的联系尤其密切,因为这些疾病的特点是蛋白异常聚集(43)。NRF2在蛋白质病中具有保护作用的令人信服的证据已经在阿尔茨海默病(AD)模型的淀粉样病变(153,154)、tau病变(155)或两者皆有(40, 156, 157)以及帕金森病(PD)模型的α-突触核蛋白病变(158-160)中得到证实。在人类,APP受损和tau损伤神经元NRF2及其靶蛋白p62 / SQSTM1表达增加(40,161)。这些发现与最近报道的NRF2上调巨自噬(40)和分子伴侣介导自噬(39)的基因表达作用是一致的,这是清除APP、tau和α-突触核蛋白的两种必要机制。NRF2上调保护神经元免受突变型α-突触核蛋白和leucinerich重复激酶2(LRRK2)的毒性(162),LRRK2也导致与错误折叠蛋白质聚集相关的神经变性(163)。奇怪的是,尽管NRF2激活增加了α-突触核蛋白的降解,但错误折叠的弥漫性LRRK2被隔离到包涵体中。
NRF2在星形胶质细胞和小胶质细胞中的作用尤其重要,因为它们在大脑中表达水平最高(图2b)。通过使用带有ARE驱动的碱性磷酸酶报告基因的转基因小鼠,我们发现星形胶质细胞在几种神经变性模型中对NRF2的激活反应高度敏感(164),这表明NRF2通过对即将死亡的神经元提供代谢和GSH前体而在星形胶质细胞的培育作用中具有相关性(46, 165)。小胶质细胞作为大脑的免疫应答细胞,在NRF2介导的应答中也起着至关重要的作用(166, 167)。在帕金森毒素甲基-4-苯基-1,2,3,6-四氢吡啶致小鼠PD模型中,NRF2可调节小胶质细胞动力学,从而降低COX2、NOS2、IL-6和TNF的生成,并增加多种抗炎标志物的水平(55, 168)。tau损伤神经元激活小胶质细胞似乎至少部分地通过与分形趋化因子相互作用有关。在小鼠和人类,tau损伤神经元释放出分形趋化因子,抑制邻近的小胶质细胞中GSK3并导致NRF2蛋白的增加。反过来,NRF2减少TNF和IL-6的释放,并参与小胶质细胞的重编程,以达到愈合伤口(161)。
一些NRF2靶基因,包括HMOX1、NQO1、GCLM和SQSTM1,在AD和PD脑中上调(159,169-171)。通过对NFE2L2启动子中3个SNP的功能单倍型与疾病风险之间关系的遗传分析,证明了这种上调的相关性。在肌萎缩性脊髓侧索硬化症(ALS)中,一种保护性的单倍体等位基因与疾病发作延迟4年有关(172),但另一项研究没有发现明显的关联(173)。在AD中,另一个单倍型变异与早2年发病有关(76)。已经报道了PD的更详细证据。最初,保护性单倍型与瑞典队列中疾病发作延迟相关,并且波兰队列PD患者的风险降低(77)。之后,这些研究结果在四个独立的欧洲病例对照研究中得到了重复(174),但台湾人口却没有(175),这表明存在种族和/或环境差异。NRF2的药理激活也有望用于治疗亨廷顿病(Huntington disease, HD)和弗里德雷希共济失调(Friedreich ataxia, FRDA),对于这两种疾病来说,氧化应激和炎症是重要的病理驱动因素。在小鼠、来自HD和FRDA患者的模型生物体和培养细胞中进行的研究报告了NRF2信号通路受损(53,176 - 178)以及药理学NRF2激活剂的有益作用。因此,三唑衍生物对NRF2的激活抑制了HD原代小鼠小胶质细胞和星形胶质细胞以及来自HD患者的培养单核细胞促炎细胞因子的释放(53),并且在离体HD大鼠皮质纹状体脑切片和HD果蝇模型中具有神经保护作用(179)。在HD、DMF和三萜类化合物的小鼠模型中,CDDO-乙基酰胺和CDDO-三氟乙基酰胺改善了动物的脑病理和行为(180,181)。用SFN和环状氰基酮TBE-31或RTA-408处理改善了FRDA小鼠模型成纤维细胞、小脑颗粒神经元以及FRDA患者成纤维细胞的线粒体功能和抗氧化能力(182,183)。总而言之,现有证据表明NRF2的适度激活对大脑具有神经保护作用。
NRF2-KEAP1信号通路和癌症
据报道,NRF2具有抗肿瘤和促肿瘤作用。在这里,我们简要总结了目前对NRF2在癌症中作用的理解,并参考了最近在癌症背景下讨论NRF2的全面综述(12)。
在非恶性细胞中,NRF2激活对氧化剂诱导的遗传损伤以及化学和物理致癌物具有抵抗性,这是由于NRF2激活增强了机体的防御能力,包括抗氧化(184)、放射性保护(114)以及加速DNA损伤剂的生物转化和清除(185)。此外,NRF2转录反应的激活似乎将ROS水平维持在低于对肿瘤发生关键蛋白质发送信号所必需的水平,例如磷脂酰肌醇-4,5-二磷酸3-激酶(PI3K)、丝裂原活化激酶、缺氧诱导因子1和NF-κB(186)。
相反,在肿瘤发展的早期阶段,选择具有NRF2组成型激活的癌细胞作为能够适应恶劣微环境、化疗、放疗或高内源ROS水平的方式。此外,在快速增殖的细胞中,NRF2通过增强核苷酸(26)和氨基酸(187)的生物合成来支持中间代谢,但也会产生代谢失衡和对谷氨酰胺酶的依赖,这可能用于治疗(188,189)。
KEAP1的体细胞功能缺失突变或NFE2L2的功能获得突变在非小细胞肺癌和其他一些环境因素是重要病因的癌症中很常见(190)。癌症基因组图谱(TCGA)数据库中的NRF2突变综合目录在10364例中识别出226个独特的NRF2突变肿瘤,其中33种肿瘤类型中有21种发生了功能获得性NRF2突变(191)。这种突变的频繁发生表明NRF2抑制剂在癌症治疗中的潜在益处,从而引发对此类药物的研究(Box 1)。这些NRF2抑制剂代表了药理学的一个完全开放的领域,目前处于概念验证研究的水平。NRF2抑制的替代方法包括靶向依赖NRF2和/或在KEAP1突变癌细胞中选择性表达的蛋白。例如谷氨酰胺酶(188, 189)和NR0B1的抑制剂,NR0B1是一种非典型的孤儿核受体,在多聚体蛋白复合物中调节KEAP1突变细胞的转录(192)。然而,值得注意的是,目前尚无证据表明单单KEAP1或NFE2L2突变能够引发恶性转化,与致癌驱动因子共发生似乎是必要的,PI3K通路改变与恶性转化的相关性最强(193)。
最能说明问题的是,KEAP1亚等位基因敲低的NRF2全基因组上调小鼠不会自发形成肿瘤(194, 195),而小鼠肺中同时缺失Keap1和PTEN则会促进腺癌的形成(195)。重要的是,由此产生的肿瘤具有免疫抑制微环境的特征,可通过免疫检查点抑制剂进行治疗(195)。很明显,NRF2基因激活是肿瘤发生的重要因素,因为癌基因(KRASG12D)激活啮齿动物NRF2信号可以增加肿瘤发展(196),而KEAP1基因敲低(也导致NRF2信号激活)强烈阻碍NOTCH1驱动的肿瘤发生(197)以及紫外线辐射介导的皮肤癌发生(51, 198)。值得注意的是,在后一种模型中,同时丧失NRF2可以消除KEAP1敲低对皮肤癌发生的保护作用(199)。 NRF2的这种两面作用得到广泛的实验证据支持,在这些实验证据中,NRF2的激活增强了抗肿瘤免疫,以防止肺癌的发生,但只有在肿瘤发生后才会加速恶性生长(200)。此外,与野生型同窝小鼠相比,接受化学诱导致癌作用的NFE2L2敲除小鼠表现出肿瘤数量增加(启动增强)但体积小得多(进展受损)(201)。因此,低NRF2活性促进癌发生的起始,而持久的组成型高NRF2活性可以驱动癌症进展和对治疗的抗性(202)。
NRF2调节剂的临床开发
富马酸酯。富马酸酯代表一组NRF2激活剂,工业上对此进行了大量的研究。临床最成功的例子是DMF(化合物1,图3a),它于1994年被批准用于治疗银屑病,并且基于其在MS的EAE小鼠模型中的功效(203),于2014年被Biogen(Tecfidera)重新用于治疗复发-缓解型MS。在发现NRF2之前,对啮齿动物的早期研究报道DMF是NRF2转录靶点GST和NQO1的强效诱导剂(204),后来发现DMF代谢物富马酸单甲酯(MMF;化合物2,图3a)与KEAP1中的C151反应,激活NRF2(203)。自从发现DMF的诱导剂活性以来,安全性一直是一个优先考虑的问题,并注意到获得显著酶诱导剂所需DMF浓度对啮齿动物具有良好的耐受性(204)。DMF在两个III期试验中表现出良好的安全性和耐受性(205, 206),自其商业化以来,它已成为近年来最成功的新药之一。然而,Tecfidera的一个缺点是出现轻度至中度腹痛、潮红、腹泻和恶心。尽管可以控制这些不良反应,但是更严重的症状是在治疗开始时白细胞计数低的患者发生白细胞减少症。事实上,用DMF治疗的动物神经系统中粒细胞的水平远低于未接受该药物治疗的动物(207)。该作用与NRF2无关,但很可能是由于羟基羧酸受体的活化,因为缺乏该蛋白质的小鼠具有高水平的粒细胞,无论它们是否用DMF处理(207)。这些观察结果强调了仔细选择治疗纳入标准的重要性。
如上所述,DMF在体内代谢为MMF,通过在C151处形成加合物使KEAP1失活(203)。由于这种代谢转化,一些生物制药公司正在开发具有缓慢和持续释放MMF的化合物,与DMF相比,其显示出改善的生物利用度和更少的副作用(表1)。Alkermes(被Biogen收购)开发了富马酸迪罗西酯(BIIB098,以前为ALK8700,化合物3;图3a),一种具有降低GI副作用的MMF前药,现在正在进行MS的III期临床试验(NCT03093324)。 XenoPort(由Arbor Pharmaceuticals收购)开发了富马酸富马酰胺,一种MMF前药(XP23829,化合物4,图3a)。与DMF相比,XP23829在临床前模型中具有更高的溶解度和渗透性,口服给药后更大的吸收,改善功效和降低GI副作用,并且现在处于斑块状银屑病的II期临床试验中(NCT02173301)。Catabasis制药公司正在开发另一种MMF和二十二碳六烯酸(CAT4001)的化学连接偶联物,与单独的生物活性分子相比,这种偶联物具有增强细胞靶向性、有效性、安全性和耐受性的潜力。特定的酶释放细胞内的两个组件,同时调整多个生物目标,包括细胞和动物模型中的NRF2和NF-κB, 并显示出治疗神经退行性疾病(如FRDA和ALS)的前景。V ClinBio正在开发一种类似的技术,用于治疗MS和银屑病的MMF和二十碳五烯酸(VCB101,化合物5,图3a和VCB102)的结合物(表1)。
萝卜硫素(SFN)。SFN一种广泛应用的天然亲电NRF2激活剂 (化合物6,图3a)。该化合物最初由Paul Talalay和Yuesheng Zhang作为十字花科植物提取物NRF2靶酶NQO1的主要诱导物分离得到的(208),并与KEAP1的C151相互作用(14,209)。值得注意的是,SFN可通过血脑屏障(55),并在许多神经系统疾病的临床前模型中具有保护作用(210)。在一项针对患有自闭症谱系障碍年轻男性的双盲安慰剂对照临床试验中,口服富含SFN的西兰花芽提取物胶囊,通过异常行为检查表和社会反应量表评估,在行为方面改善显著;这些量表上的总分数在停止治疗后逆转至治疗前水平(211)。三日龄的西兰花芽富含葡萄糖苷,一种SFN前体分子,可被植物黑芥子酶水解,释放出SFN和葡萄糖(212)。类似的β-硫葡糖苷酶存在于肠道微生物群中;因此,释放的SFN实际水平高度依赖于饮食习惯和微生物组成,受抗生素治疗的影响(213)并表现出昼夜节律(214)。并且,无论是芽苗提取物还是高度纯化的SFN,已有超过500名受试者接受超过25,000剂量,证明了具有高度安全性。目前有超过20个正在进行的临床试验。
考虑到天然化合物知识产权的相关问题、SFN在室温下不稳定以及需要精确控制剂量,Evgen Pharma开发了SFN的药物形式,SFX-01(化合物7,图3a) 。 SFX-01是化学合成的SFN,包裹在环糊精中,形成稳定的固体形式的丸剂或胶囊,具有良好的生物利用度。SFX-01目前正在用于蛛网膜下腔出血(NCT02614742)和转移性乳腺癌(NCT02970682)临床试验(表1)。Evgen还有一系列基于SFN的新型类似物,这些类似物正处于临床前评估阶段。此外,许多营养保健品公司正在生产含有NRF2诱导剂的制剂,包括SFN,具有不同程度的标准化。其中一个例子是来自Nutrinov实验室的Prostaphane,它是一种稳定的游离SFN,从花椰菜种子中提取,在前列腺癌的安慰剂对照临床试验中显示了在根治性前列腺切除术后控制生化复发的优势(215)。SFX-01和Prostaphane的生物利用度和效力等同于稳定性较差的SFN(216)。另一个例子是胶囊膳食补充剂Avmacol(Nutramax Laboratories),含有来自精细研磨的花椰菜种子的萝卜硫苷和冷冻干燥的西兰花芽粉末以提供黑芥子酶。该补充剂目前正在临床试验中用于调节与自闭症谱系障碍、精神分裂症和环境污染有关的疾病症状和/或生物标志物(217)。
氰基酮三萜。越来越多的证据表明,富马酸酯、SFN以及与KEAP1共价结合的其他小亲电试剂可以从更大的支架结构中受益,以赋予更好的选择性(和可能的效力)和更可控的药代动力学和/或药效学特性。含五环迈克尔受体的氰基酮三萜类化合物的开发可满足这些要求。这些五环三萜类化合物最初由Michael Sporn、Gordon Gribble和Tadashi Honda开发,来自天然产物齐墩果酸(218, 219),这些五环三萜类化合物是迄今已知最有效的亲电子NRF2激活剂(220),由KEAP1中的C151检测,目前正在由Reata Pharmaceuticals和Kyowa Hakko Kirin进行临床开发。
两种临床上先进的化合物,bardoxolone methyl(BARD;化合物8,图3a)和omaveloxolone(化合物9),能使代谢正常化、增加线粒体能量产生、增强细胞抗氧化能力和降低ROS水平(221)(表1)。在临床前研究中,BARD或类似物已被证明可通过减少炎症、纤维化和氧化应激以及增加肾小球的过滤表面积来改善肾功能(222, 223)。通过修改选择标准以排除有早期心力衰竭迹象的患者(B型利钠肽升高),BARD目前正在临床试验中测试几种与慢性肾病(CKD)相关的罕见病症,大多数没有经批准的治疗方法,包括Alport综合征(NCT03019185)、常染色体显性遗传性多囊肾病、免疫球蛋白A(IgA)肾病、1型糖尿病和局灶性节段性肾小球硬化(NCT03366337),其中促炎和促纤维化过程造成肾小球硬化和肾功能受损(224)。BARD治疗有可能延缓或预防肾小球滤过率下降,而肾小球滤过率下降会导致Alport综合征和其他罕见形式CKD患者需要透析或移植。因此,在患有T2DM和CKD患者的II期临床试验中(NCT02316821),BARD治疗可使直接测量肾小球滤过率的统计学显著增加,并且在排除具有液体潴留风险患者后,耐受性良好。在这些结果的基础上,Kyowa Hakko Kirin在患有G3期或G4期CKD的糖尿病患者中,在日本SAKIGAKE指定系统的支持下,开展了III期临床试验(NCT03550443)。
此外,BARD正在用于研究肺动脉高压(PAH)(NCT03068130)和结缔组织病(CTD-PAH)引起的PAH(NCT02657356),这是一种导致心力衰竭和死亡的严重和进行性疾病。CTD-PAH与特发性病因之间的差异很大程度上归因于炎症、自身免疫和系统性血管病变之间复杂的相互作用,这些相互作用促成了结缔组织疾病的发病机制。由于NF-κB的上调,CTD-PAH患者的炎症加重(225),对现有血管扩张剂治疗的反应性低于特发性PAH患者,预后较差(226)。在II期研究中,对CTD-PAH患者进行了BARD测试,目前正在III期研究中进行测试。Omaveloxolone目前正在FRDA患者(NCT02255435)的II期临床试验中进行测试,这是一种遗传性神经肌肉疾病,其NRF2被下调(176-178)。值得注意的是,目前尚无批准治疗FRDA的疗法(227)(表1)。
硝基脂肪酸。脂肪酸的硝化衍生物(NO2-FAs)是内源性信号介质,在代谢和炎症疾病的临床前动物模型中具有抗炎和抗纤维化活性(228)。硝基烯烃基团赋予其β-碳亲电性,促进与亲核试剂如半胱氨酸的可逆NO2-FA迈克尔加合物的快速形成,这种改性称为硝基烷基化(22, 230)。已经表明,硝基油酸(NO2-OA; 9-硝基十八碳-9-烯酸)与KEAP1中的半胱氨酸反应,包括C273和C288,从而激活NRF2(231,232)。该反应的可逆性阻止了稳定的NO2-FA硫醇加合物积累的可能性,这可能导致细胞毒性(219,230,233)。实际上,I期安全性评估证明了先导化合物CXA10(10-NO2-OA; 10-硝基-十八碳-9-烯酸,NO2-OA的特异性区域异构体)(化合物10,图3a)在药理活性剂量下对人体是安全的(234)。目前,Complexa正在开发CXA10作为局灶性节段性肾小球硬化和PAH的治疗方法(234)(表1)。最近报道了来自α-生育酚的硝基烯烃合成和生物学评价(235)。
羟胺。一个特殊的挑战是提供可以通过血脑屏障的NRF2激活剂。给药途径(注射、口服或局部)对特定疾病也至关重要。Othera Pharmaceuticals(现由Colby Pharmaceutical收购)正在开发一种具有抗氧化特性的二取代羟胺(OT551),局部应用,通过靶向KEAP1抑制氧化应激和疾病相关炎症(化合物11,图3a)。在临床前研究中,该化合物的眼用溶液保护视网膜色素上皮细胞和光感受器免受氧化损伤和炎症反应,并且一项关于年龄相关性黄斑变性的II期试验已证明可有效预防视力丧失的进展(NCT00485394)(表1)。
TFM735。一种高通量筛选报告子策略评估了包含融合到LacZ (NRF2d-LacZ)236的NRF2氨基末端区域嵌合蛋白的稳定性,该策略确定TFM735为先导化合物(化合物12,图3a)。TFM735以C151依赖性方式激活NRF2,并抑制受刺激的人外周血单核细胞中IL-6和IL-17合成以及小鼠EAE进展(237)。该化合物目前由Mochida Pharmaceuticals进行治疗MS的临床前开发(表1)。
NRF2-KEAP1蛋白-蛋白相互作用抑制剂。近年来,非亲电性非共价化合物的发展受到关注,这些化合物可以直接干扰KEAP1和NRF2之间的PPI(238),或者干扰KEAP1和CUL3之间的PPI(239)。通过直接干扰KEAP1和NRF2或CUL3之间的相互作用,化合物的作用方式不需要共价结合组分。该方法具有潜在的优点,包括探索NRF2诱导物的新化学型的范围,由于与KEAP1的不同相互作用模式导致的不同药效学以及由于半胱氨酸非依赖性结合机制导致的不同脱靶效应谱。有证据表明,亲电子和非亲电子(PPI)KEAP1抑制剂的生物学效应谱不同,一个例子是非亲电试剂诱导线粒体自噬的能力,与SFN和DMF等亲电试剂相比;这表明两种化合物类别之间药理活性和治疗效用的潜在差异。
PPI抑制剂的设计是以KEAP1的Kelch结构域的晶体结构可用性为指导的(241-243)(图3b)。已经报道了几种类型的PPI抑制剂;根据制药行业投入开发的主要化学类别如图3b所示。萘双磺酰胺化合物起源于Biogen的高通量筛选(244),随后由中国药科大学Jiang等人提供化合物13(CPUY192018)和类似物(245)。该双羧酸化合物是KEAP1的高亲和力配体,在低微摩尔浓度下诱导ARE依赖性基因的表达。Astex和葛兰素史克还从基于X射线晶体学碎片的筛选程序中鉴定了含磺酰胺的铅化合物。在这种情况下先导化合物的例子是化合物14,KEAP1-NRF2 PPI低纳摩尔抑制剂,它能增加COPD患者上皮细胞中NRF2靶基因NQO1的表达。该化合物能够在静脉内给药后减轻大鼠的肺部炎症,并有效抑制臭氧诱导的支气管肺泡液中白细胞的积累并恢复GSH浓度(246)。来自该系列的化合物已经在几种模拟肺病特征的氧化应激动物模型中显示出功效,并且目前正在开发用于临床评估。已经开发了另外两类不是磺酰胺的单酸性PPI抑制剂。第一类是由胡等人在Rutgers开发(247),由Evotec和UCB Pharma详细描述(248),包括四氢异喹啉(例如化合物15),它们是KEAP1-NRF2 PPI的低微摩尔抑制剂。第二类是由Toray Industries和RIKEN开发的,并加入了恶二唑基序;化合物16是PPI抑制剂,具有微摩尔范围内的结合活性(249)。突出显示的化合物或其类似物已与KEAP1 Kelch域共结晶;图3b显示了化合物13-16所获得的结合位点亚袋的占用情况,并为这些配体未来的精炼提供了思路。
C4X Discovery利用计算化学、配体核磁共振光谱和蛋白质结晶学方法以及Keapstone利用基于结构的药物设计方法联合开发了新的PPI抑制剂。学术团体也在为非亲电NRF2诱导物研究做出贡献(250,251)。
如何使PPI抑制剂具有适当的药物代谢和药代动力学特性,用于外周神经系统和中枢神经系统,仍然是一个重大挑战,后者是一个需要具有强安全性NRF2激活剂用于长期给药等条件的领域,如AD和PD。此时,非亲电性PPI抑制剂改善靶向选择性的潜在优势被当前化合物相对较高的分子质量、阻断较大KEAP1-NRF2界面的要求以及极性官能团赋予KEAP1紧密结合亲和力的需要所抵消。因此,目前通过体外研究鉴定的许多原型化合物表现出较差的吸收、分布、代谢和排泄性质。例如,萘双磺酰胺(化合物13)对DSS诱导的小鼠结肠炎具有保护作用,在低微摩尔范围内激活NCM460培养细胞中NQO1表达,并在体外与纳米级Kd结合,说明蛋白质结合与生物活性之间的相互抵消(252)。
大多数PPI抑制剂的体内评价都集中在外周炎症。例如,化合物NK-252(化合物16的类似物)被Toray Industries鉴定为相当弱的KEAP1抑制剂,但对H2O2诱导的细胞毒性具有保护作用并且降低了胆碱缺乏的l-氨基甲酸盐饮食大鼠(NASH模型系统)的纤维化评分(253)。
用KEAP1非依赖性药物靶向NRF2。越来越多的证据表明,NRF2除了KEAP1所发挥的作用外还表现出多层调节,如转录(196)、表观遗传(254,255)、共价蛋白修饰(258,257),以KEAP1非依赖性方式进行蛋白酶体降解(258-261)以及NRF2二聚体伴侣的靶向结合ARE序列的调节(262,263)。在药理学发展水平上,ARE介导的基因调控的KEAP1非依赖性机制涉及调节转录抑制因子BACH1(广泛复合物,tramtrack、bricàbrac和cap'n'collar同源体1),该机制抑制NRF2调控基因亚基的失活,尤其是HMOX1(28)。vTv 疗法现在正在分析该亚基是否足以引起治疗用处。他们开发了一类新的非亲电子小分子,可以独立于KEAP1抑制BACH1与某些ARE驱动基因的结合(264)。
挑战与思考
药效学评价。考虑到NRF2的半衰期很短,即使在药物诱导稳定后,也需要通过间接指标来推断其在病变器官中的激活情况,从而确定最合适的治疗方案。一种可能性是分析可接触的细胞或组织(如外周血单核细胞、鼻腔灌洗液细胞、脱落的膀胱细胞、颊细胞和皮肤)中的药物分布和NRF2基因表达特征,希望NRF2的转录组特征反映了其他组织(如肺,肝和脑)的局部参与(265-267)。群体不可避免地暴露于有害环境化学物质的情况下,尿液中相应代谢物的水平已被用作NRF2介导的药效学作用生物标志物(268,269)。
NRF2通路的激活表现为依赖于时间、组织、剂量、发病时间和持续时间的差异基因表达。表2显示了NRF2调控基因产物代表性亚群的半衰期。由于其中许多蛋白质的半衰期相当长,其生物化学活性的持续时间预计将远远长于NRF2稳定的时间间隔或其药理激活剂的存在,后者通常在数小时内被清除。因此,NRF2活化分子的药效学观察时间比NRF2水平要长得多,且与血浆药物浓度不一致。因此,NRF2激活剂TBE-31(一种与上述三萜类化合物密切相关的三环氰基酮)在小鼠皮肤和血浆中的半衰期为10小时(51,270),而NRF2依赖的NQO 1诱导蛋白在最后一剂局部应用药物3天后仍可在皮肤中检测到(51)。因为NRF2激活剂导致“刺激产生或抑制控制测量效果的因素”,所以“间接药效学反应”模型可能是用于测试NRF2激活剂药效学走向临床应用的更合理方法(272,272)。
NRF2活化药物在相关组织中的浓度和分布,以及它们的溶解度、细胞通透性、代谢稳定性和蛋白结合,也将在NRF2活化程度中发挥重要作用,最终达到预期的长效药效学效果。因此,优化临床有意义的药物暴露需要通过一个假定的NRF2激活剂分析剂量依赖性基因调控。此外,NRF2靶向参与研究必须考虑患者的年龄和表现状态,因为在年龄较大和健康状况较差的受试者中,药物激活NRF2或促进“适应性反应”的能力似乎有所下降。最近在DMF复发缓解型和多发性硬化症患者的临床试验中发现,NRF2转录靶NQO1的诱导程度与患者年龄呈负相关, 在4 - 6周高NQO1蛋白含量的患者在开始治疗后的第二年更有可能没有疾病活动的证据(267)。
药物选择性。KEAP1中高活性半胱氨酸的鉴定对于理解亲电化合物与泛素连接酶底物适配器共价作用的机制至关重要。结构信息表明,某些作用于C151的药物可能会干扰与CUL3-RBX1的相互作用(273, 274),从而使KEAP1库被结合的NRF2饱和,并允许新合成的NRF2逃避降解: 这是用亲电试剂n -碘乙酰- n -生物素己二胺模型证明的(275)。其他化合物可能会修饰KEAP1,使其不再能够在转录因子内的高亲和力和低亲和力位点与NRF2相互作用。值得注意的是,一些亲电的NRF2激活剂,如环氰烯酮,以共价形式与硫醇结合,但不形成不可逆的加合物(276,277)。因此,它们结合了不可逆共价药物(即高效力和持续的靶点参与)和可逆非共价药物(即缺乏永久性修饰,因此可能破坏或免疫原性其蛋白靶点)的理想特性(278)。这类分子的一个潜在缺点是,它们可能与KEAP1以外的蛋白质中对氧化还原敏感的半胱氨酸发生反应,从而影响其功能。巯基反应性分子的特异性问题表明,对于每种化合物,需要确定在其他蛋白质中不存在硫醇修饰的情况下允许治疗相关KEAP1修饰的窗口(图1b)。KEAP1内的靶半胱氨酸以及其他含巯基的非靶蛋白的这种分层呈现被称为“半胱氨酸代码”(279),并且受诱导剂的化学和剂量影响(280)。
KEAP1-NRF2 PPI抑制剂的一个重要潜在优势是提高了靶点的选择性。然而,对于任何小分子,不能完全排除脱靶效应。此外,KEAP1靶向NRF2以外的蛋白进行泛素化,这些蛋白也可能受到影响(281)。PPI抑制剂的脱靶选择性已被评估的几种类型之一是强效单酸性磺胺类;例如,化合物14似乎对KEAP1具有相当的选择性,在葛兰素史克公司(GlaxoSmithKline)增强的体外实验交叉屏幕面板中显示出适度的活性,以识别潜在的脱靶效应(246)。
动物模型。阻碍大多数慢性病药物开发成功的一个问题是动物模型中人类病理的不完全可复制性(288)。这一问题可能与人类退行性疾病缺乏一个非常重要的标志有关,即与NRF2相关的体内平衡功能的逐渐丧失。事实上,尽管在人类仍有许多工作需要做,但动物研究证据表明,NRF2活性随着年龄的增长而下降,并且NRF2的药理学或遗传学上调可延长寿命并改善健康状况(283, 284)。慢性疾病动物模型缺乏“NRF2变量”,这在AD小鼠模型中得到了很好的证明。目前的模型是基于这样的假设,即有毒的APP或tau突变蛋白在原本健康的动物背景下的表达复制了人类的病理。尽管这些模型提供了关于蛋白病的发病和进展的有价值信息,但是它们未能将治疗成功应用于人类。这种失败的原因可能与这些模型不考虑与NRF2相关稳态功能的疾病相关性下降这一事实有关。更复杂的临床前模型将需要一种反向翻译方法,这种方法不仅要在动物身上复制特定疾病的解剖病理特征,而且要复制受损的氧化还原、炎症或蛋白稳态。因此,NRF2敲除小鼠与AD患者甚至老年人有许多共同改变(156)。最近的研究表明,与野生型动物相比,喂食高脂肪食物的NRF2敲除小鼠表现出更大的神经血管功能障碍、血脑屏障破坏、神经炎症、淀粉样基因表达和认知能力下降,这与随着年龄增长而发生的许多表型变化类似(285)。引入低NRF2表达作为降低稳态反应的变量,可能有助于改进现有的慢性病模型,以获得更好的治疗效果。考虑到随着衰老NRF2活性的缓慢下降,杂合的NFE2L2 +/-小鼠可能是复制基础表达减少和药理学上调的合适模型。
癌症风险。从药理学角度来看,有必要确定NRF2水平升高超过致癌安全阈值的风险有多大(图4)。促进NRF2在癌细胞中稳定性的体细胞突变影响与NRF2的药理学激活引起的影响存在显著差异。因此,KEAP1和NFE2L2的体细胞突变导致NRF2活性升高而不受限制,这与NRF2激活剂的药理作用引起的脉冲激活完全不同。动力学特征,如曲线下的振幅和面积,反映NRF2信号的强度和持续时间,在基因激活设置和药理学手段方面有很大差异(202, 286)。此外,KEAP1基因丢失或失活突变似乎与该蛋白的药理学抑制作用不一样。因此,KEAP1丢失导致的NRF2活性以及E3连接酶适配器的几个致癌靶标(包括IKKβ和B细胞淋巴瘤2,bcl-2)增加,可能至少是恶性转化的部分原因。因此,在具有功能性KEAP1的一组细胞系中,成熟的NRF2激活剂,即BARD类似物RTA-405,增加NRF2但不增加IKKβ或BCL-2水平,并且不赋予肿瘤细胞生长或存活优势(287)。
虽然目前NRF2激活剂的癌症风险不能被忽视,但令人鼓舞的是,DMF III期试验荟萃分析显示安慰剂和DMF治疗组之间的癌症发生率没有差异(288)。 同样重要的是要注意,一些NRF2激活药物如果具有抗肿瘤作用的其他靶标可以降低这种风险。因此,最近发现DMF抑制GAPDH(289),并且这种抑制可能导致高度糖酵解的KRAS或BRAF突变癌细胞的能量危象,从而阻止肿瘤发展,如维生素C(290)。
展望
许多慢性疾病的标志是丧失稳态,如氧化还原信号、代谢灵活性、炎症控制和蛋白质稳态。这些复杂疾病的多因素性质可以通过对转录因子NRF2的单次靶向激活,产生有益的、全面的、多靶点和持久的细胞保护作用。尽管KEAP1-NRF2系统的特殊性使得这种方法在监测靶向参与和脱靶效应方面特别具有挑战性,但生物制药公司正在缓慢而稳定地开发针对NRF2主要调控因子KEAP1的药物。此外,需要明确排除增加癌症风险的担忧。在未来几年,正在进行的临床试验无疑将在回答这些问题方面取得重要进展。
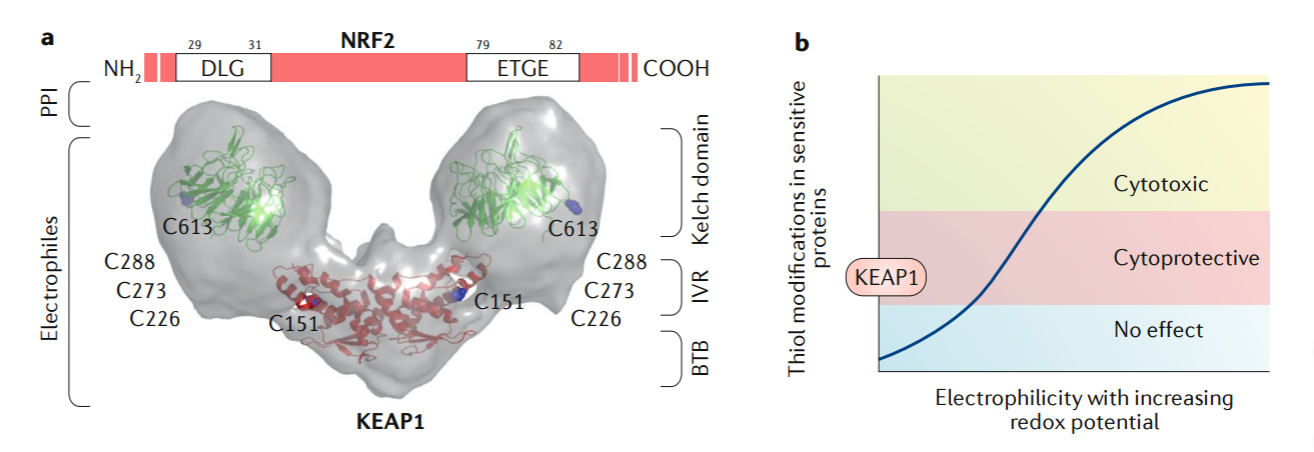
图1:KEAP1对NRF2的调控及其药理靶标。
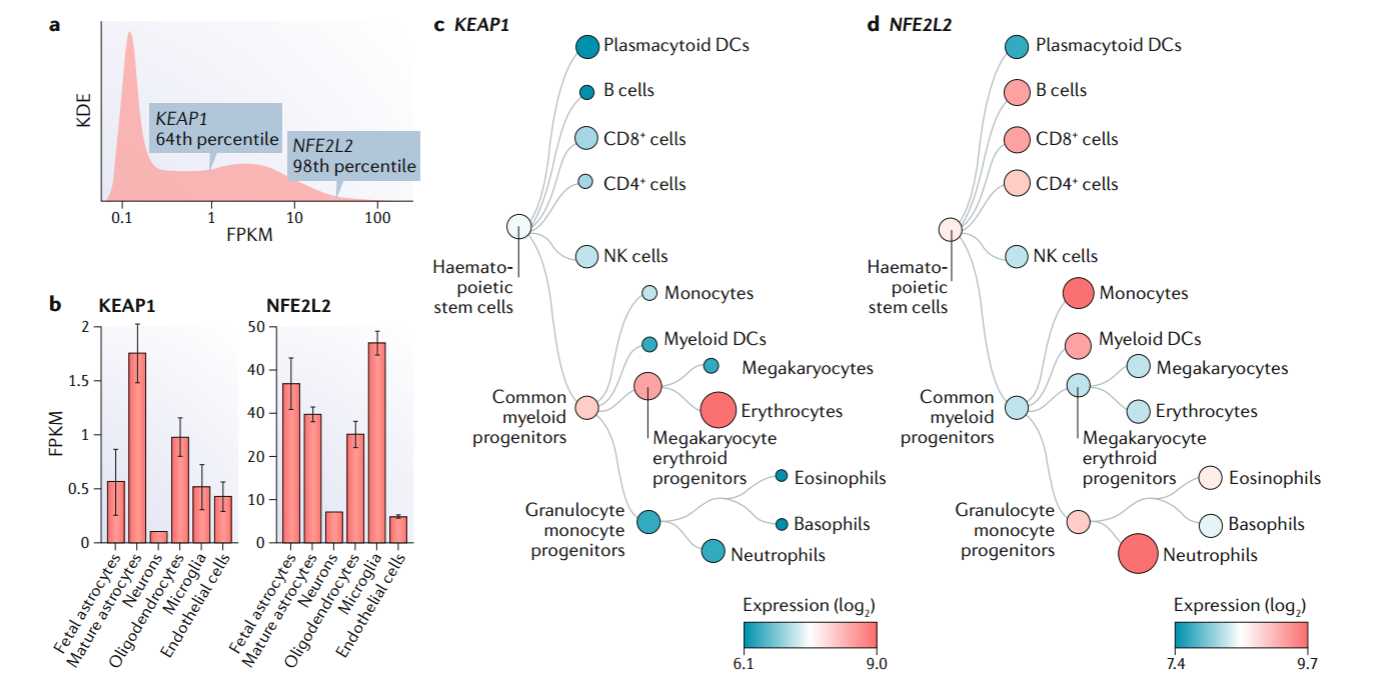
图2:KEAP1和NFE2L2 mRNA在人脑和血液中的表达。
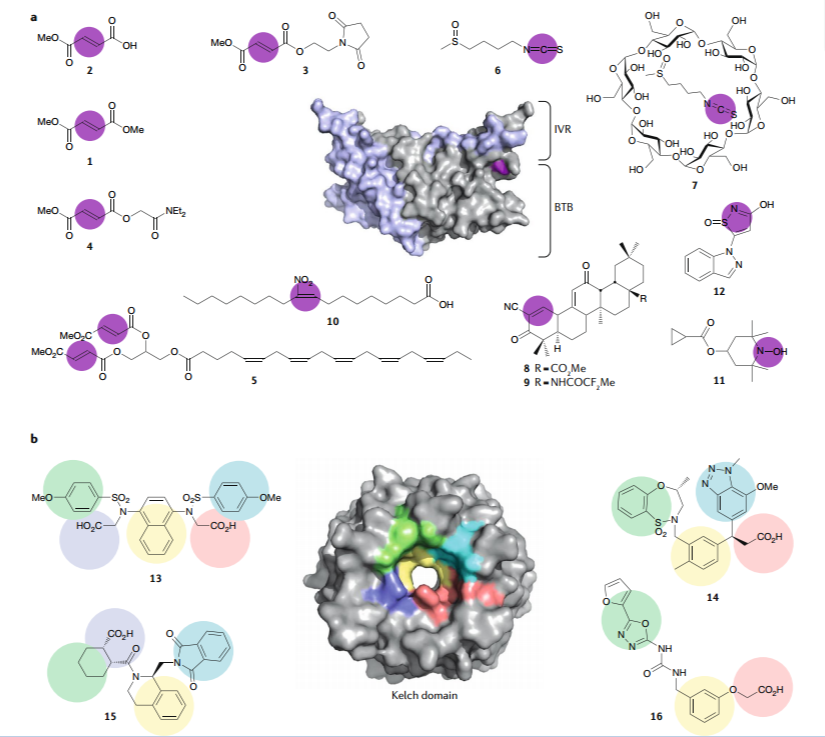
图3:KEAP1结构域的晶体结构。
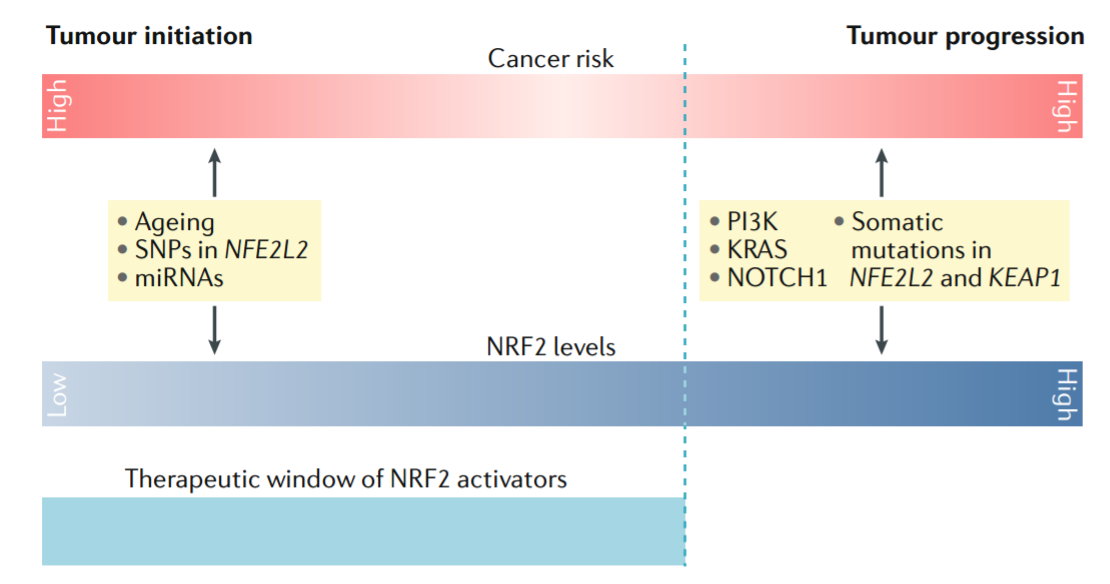
图4:根据NRF2的状态调节癌症风险。
摘要:转录因子NF-E2 p45相关因子2(NRF2;由NFE2L2编码)及其主要负调节因子,E3连接酶受体Kelch样ECH相关蛋白1(KEAP1)是维持氧化还原、代谢和蛋白质稳态以及炎症调节的关键。因此,激活NRF2可对多种疾病起到细胞保护作用,包括肺和肝的慢性疾病、自身免疫病、神经退行性疾病和代谢性疾病以及癌症启动。一种NRF2激活剂已获得临床批准,并且基于半胱氨酸传感器KEAP1的几种亲电修饰剂及其与NRF2相互作用的抑制剂目前正在临床开发中。然而,在靶向特异性、药效学特性、疗效和安全性方面仍存在挑战。
前言
转录因子NF-E2 p45相关因子2(NRF2;由NFE2L2编码)于1994年作为人类cap 'n' collar(CNC)碱性区域亮氨酸拉链转录因子家族成员被发现(1)。随后的研究,包括NRF2敲除小鼠的产生,确定NRF2调控约250个基因的表达,调控的基因在其启动子调节区中含有增强子序列,称为抗氧化应答元件(ARE)(2, 3)。这些基因编码一个合作酶网络,参与内生和外生生物转化反应、抗氧化代谢、碳水化合物和脂类的中间代谢、铁的分解代谢、蛋白质降解和炎症调节(4)。通过这个转录网络,NRF2能够协调多方面应对各种形式的应激,维持稳定的内部环境。
1999年,Kelch样ECH相关蛋白1(KEAP1)被鉴定为NRF2的抑制因子(5),又过了5年才被报道为E3泛素连接酶底物适配器,靶向NRF2进行蛋白酶体的快速降解(6-8)。因此,KEAP1确保在无应激条件下,NRF2是低丰度的蛋白质,具有仅约15-40分钟的有限半衰期,这取决于细胞类型。然而,重要的是,KEAP1含有几个高活性的半胱氨酸,经亲电子分子修饰后,可防止其靶向NRF2进行蛋白酶体降解,从而导致NRF2蛋白在氧化还原应激下快速核聚集,并在与小肌肉筋膜纤维肉瘤癌基因同源蛋白(sMAF)二聚化后,通过NRF2-sMAF诱导含有ARE的基因(9,10)。
现在人们普遍认识到,由于NRF2-靶基因编码的蛋白质的多功能和全面的细胞保护作用(包括抗氧化、解毒和抗炎),因此,NRF2-KEAP1信号通路对于预防以氧化应激和炎症为基础病理特征的多种疾病至关重要。这些疾病包括代谢、炎症和自身免疫性疾病,肺、肝、肾、胃肠道和心血管系统疾病以及神经疾病(9, 11)。NRF2在这些非肿瘤性疾病中的保护作用的实验证据将在本综述的后面讨论。我们还讨论了NRF2在癌症中的作用,这是一个需要深入研究的问题(12)。
通过单粒子电子显微镜获得的KEAP1二聚体3D重建显示,该蛋白具有叉状茎结构,包括两个大球体,它们包围了中间区域(IVR)、Kelch和羧基末端结构域,而二聚化由BTB域介导(图1a)(13)。考虑到KEAP1包含许多不同且自主运作的半胱氨酸传感器(C151、C226、C273、C288和C613)(14-16),现有证据表明,NRF2和KEAP1整合了一氧化氮、Zn2+、烯醛信号传导与氧化还原信号传导一起提供了一种可能至少部分进化的机制,以允许快速适应饮食中遇到的植物化学物质。许多植物来源的亲电异种生物激活NRF2-KEAP1信号通路以引发细胞保护反应,KEAP1中的高活性半胱氨酸起直接传感器的作用(17-19)。这些传感器与亲电子试剂的高反应性使得KEAP1在没有非特异性硫醇修饰的情况下进行靶向治疗成为可能(图1b)。因此,已经描述了许多天然存在的NRF2靶基因诱导剂(20),并且新的诱导剂在不断被发现。
尽管大量学术实验室参与了NRF2激活剂的鉴定,但从历史上看,制药公司在这个令人兴奋和充满希望领域的参与度却相当低。业界之所以不愿将NRF2-keap1信号通路视为有价值的药物靶点,部分原因可能是其广泛的生物化学活性,而不仅仅是控制由NRF2直接或间接调控的抗氧化系统。NRF2激活剂的各种直接和间接效应有时被解释为非特异性效应的证据。然而,KEAP1中几种不同的基于硫醇的氧化还原开关的进化允许哺乳动物适应广泛的植物化学物质,这为改变NRF2的激活引起某些“脱靶”效应并采用更广泛视野的观念提供了理论基础。NRF2的激活产生有益的“多靶点”细胞保护作用(11)。尤其是NRF2激活的多靶点效应包括维持氧化还原信号、增强异种生物转化、控制和解决炎症、抑制糖异生和肝脂肪生成、支持蛋白稳定和抑制纤维化(9)。在这方面,值得注意的是,大多数慢性疾病,特别是老年人的慢性病,没有独特的病因学,也没有单一的病理表型,因此,它们可能通过激活NRF2的药物得到最有效的治疗。事实上,与更有针对性的治疗干预相比,NRF2激活后广泛的细胞保护机制的刺激不太可能被致病过程有效地淹没或规避。
这种对NRF2作为潜在药物靶标的广泛认可引起了制药业的兴趣,并导致对NRF2调节剂临床开发的大量投资(21)。NRF2激活剂富马酸二甲酯(DMF;目前,商标名如Fumaderm或Tecfidera)已被临床用于治疗银屑病和复发性多发性硬化症(MS)(表1),其目前的分布情况仍是其他制药公司希望达到的目标。目前,多种不同化学类型的NRF2激活剂处于临床开发的不同阶段,如靶向KEAP1半胱氨酸的富马酸衍生物、异硫氰酸硫萝卜硫素(SFN;4‐异硫氰酸甲酰丁酯)、氰烯酮三萜、硝基脂肪酸和羟胺。其他候选药物包括破坏NRF2-KEAP1蛋白-蛋白相互作用(PPIs)的非亲电化合物,以及具有keap1独立作用模式的分子。NRF2的抑制剂也在研究中,但尚未进行临床试验。本综述概述了这些候选药物,并强调了针对NRF2-KEAP1信号通路不寻常的性质和至关重要的意义。本文描述了NRF2在细胞氧化还原、代谢和蛋白稳态以及炎症调控中的生理作用,并为NRF2在非肿瘤性疾病中的保护作用提供了实验证据。评价了目前临床开发的各种小分子NRF2活性药理调节剂的优缺点。最后,强调了与NRF2调节剂开发相关的持续挑战,包括靶点特异性、药效学评估、生物利用度、有效性和安全性。
NRF2-KEAP1信号通路的生理作用
氧化还原、代谢和蛋白质稳态
自从其最初被发现参与生物转化反应,调节某些细胞色素P450氧化还原酶、结合酶和ABC转运蛋白的表达(表2),NRF2参与控制细胞内氧化还原环境的作用变得越来越明显,最终将NRF2-KEAP1信号通路识别为“硫醇驱动的总开关”用于“整个系统范围的氧化应激反应”(22)。NRF2调控参与NADPH生成的4个基因(葡萄糖6-磷酸脱氢酶、6-磷酸葡萄糖酸脱氢酶、苹果酸酶1和异柠檬酸脱氢酶1)的表达(23–26)。这种还原当量的供应进一步被大量氧化还原反应所利用,其中许多氧化还原反应也受到NRF2的调节。因此,NRF2调控氧化还原缓冲物还原型谷胱甘肽(GSH;其氧化形式为GSSG),如谷氨酸半胱氨酸连接酶、谷胱甘肽还原酶、谷胱甘肽过氧化物酶和数个谷胱甘肽s -转移酶的催化和调节亚基(4)。此外,氧化还原蛋白家族中的许多辅助蛋白,如硫氧还蛋白、硫氧还蛋白还原酶、过氧化物还蛋白和硫还蛋白,都受到NRF2的调控,提供区域活性氧生成的区域化传感和信号转导(ROS;即过氧化氢(H2O2)羟基和超氧自由基 )(27-29)。除了这些对氧化还原状态的直接影响外,NRF2还调节编码酶的基因,这些酶可阻止醌参与氧化还原循环反应和谷胱甘肽耗竭(通过NAD(P)H:醌氧化还原酶1(NQO1))或参与间接生产胆红素(血红素加氧酶1(HMOX1)和胆绿素还原酶(BVR)),是最有效的非极性生理抗氧化剂(30)。最后,NRF2通过与戊糖磷酸途径和糖酵解的相互作用,影响中间代谢,增加底物的可用性,减少线粒体呼吸链的等效物(31),并维持线粒体DNA (mtDNA)的完整性(32)。
NRF2在氧化还原改变或营养缺乏期间参与清除氧化或其他损伤的蛋白质和细胞器。因此,自噬转运蛋白泛素结合蛋白p62(由SQSTM1编码)是NRF2的转录靶点,它通过其泛素关联域(UBA)与泛素化产物相互作用,并通过其LC3相互作用的基序将其招募到自噬体中(33, 34)。此外,KEAP1与p62结合(33, 35),最近的一项研究发现KEAP1-CULLIN 3(CUL3)在其UBA结构域中的K420位点泛素化p62,从而促进p62的螯合活性(36)。在其一级结构中,p62含有ETGE样基序STGE,在被mTORC1磷酸化后产生识别位点与KEAP1对接(37)。然后,KEAP1被转运到自噬体降解,从而使NRF2积累(38)。此外,一些自噬基因似乎在其启动子区域含有序列,有报道称NRF2同时激活伴侣介导的自噬(39)和巨自噬(40,41)。NRF2在氧化条件下在蛋白质稳态中的其他作用通过未折叠蛋白质反应的激活(42,43)及其调节蛋白酶体亚基表达的能力来证明(44,45)。
至少在脑和血液中,编码NRF2的mRNA丰度高于KEAP1的mRNA(图2);相反,KEAP1蛋白的半衰期比NRF2的半衰期长(表2)。这些观察结果强调了两种蛋白质的不同转换率——KEAP1缓慢,NRF2快。此外,尽管NRF2和KEAP1普遍表达,但特定细胞类型主要负责稳态适应并呈现不同的表达水平(图2)。因此,单核细胞和嗜中性粒细胞在血细胞中表现出最高水平的NRF2,表明先天免疫系统的免疫调节作用(见下文)。在大脑中,星形胶质细胞和神经元之间的相互作用将中间代谢与氧化还原稳态结合起来,至少部分通过NRF2实现(46)。此外,神经元中的谷氨酸能神经传递导致产生由神经元GSH池控制的ROS。当该池浓度较低时,邻近的星形胶质细胞通过向神经元提供GSH前体甘氨酸、谷氨酸/谷氨酰胺和半胱氨酸来促进其恢复(47)。毫不奇怪,星形胶质细胞中NRF2水平很高(图2)。此外,与神经元相比,小胶质细胞作为单核细胞谱系的一部分表达高水平的NRF2。
控制炎症
NRF2在单核细胞和粒细胞中含量丰富(图2),提示NRF2参与了这些细胞类型驱动的免疫应答。敲除NRF2的小鼠对败血症休克高度敏感(48),并且在创伤愈合过程中表现为持续性炎症(49, 50)。而NRF2的基因或药理激活抑制了促炎细胞因子的产生(51 - 56)。在人类细胞中,KEAP1的破坏通过减弱髓系白细胞(57)和巨噬细胞(58)的炎症反应来预防败血症。特别有趣的是,NFE2L2中的一个多态性与转录活性降低相关,与炎症性肠病(IBD)(59)和慢性胃炎(60)风险增加相关。如下所示,NRF2的强抗炎活性至少可归因于三个独立的机制:氧化还原代谢的调节;与核因子-κB(NF-κB)的相互作用;对促炎基因的直接调控。
炎症与局部和全身病理水平的ROS积累增加有关,这可能损害氧化还原信号(61)。线粒体功能障碍和NADPH氧化酶不受控制的激活是炎症细胞中ROS生成升高的主要因素,线粒体ROS引起mtDNA损伤和释放,从而形成恶性循环,进一步ROS的产生和炎性小体的激活,最终导致器官功能衰竭(62)。NRF2的氧化还原调节活性是阻止炎症恶化和随后组织损伤的恶性循环中的重要突破(63)。
NF-κB和NRF2相互作用。几个NF-κB功能结合位点已确定在NFE2L2基因启动子区(64)。另一方面,NRF2抑制NF-κB转录活性。用脂多糖(LPS)或TNF处理NRF2缺陷小鼠增加了NF-κB转录的表达,表明NRF2对NF-κB的抑制作用(48)。该发现的可能解释是NF-κB抑制剂(IκB)在NRF2缺陷细胞中高度磷酸化并且经历快速蛋白酶体降解,从而减少对NF-κB的抑制(48)。此外,LPS显示同时激活NF-κB介导的快速反应和NRF2介导的缓慢反应,最初的促炎反应由NF-κB驱动,然后当NRF2活性最大时随后被抑制(65)。NRF2-KEAP1信号通路抑制NF-κB信号传导的其他机制包括NRF2和p65之间与p300竞争性结合(66)和KEAP1对IκB激酶-β(IKKβ)的降解(67, 68)。
几种巨噬细胞特异性基因含有ARE序列,因此受NRF2调节。这些包括MARCO(细菌吞噬作用所需的受体)和CD36(氧化低密度脂蛋白的清道夫受体)(69, 70)。 另一方面,NRF2以不依赖ARE的方式结合编码IL-6和IL-1β促炎基因调节区,并阻止RNA聚合酶II开始转录募集(52)。
最近的研究表明巨噬细胞代谢与NRF2引起的抗炎反应有关。线粒体代谢物亚甲基丁二酸是由三羧酸循环在促炎刺激下产生的,在巨噬细胞活化过程中会显著增加(71, 72)。反过来,亚甲基丁二酸烷基化几个KEAP1半胱氨酸传感器,包括关键C151、C273和C288残基,从而激活NRF2并抑制促炎细胞因子如IL-6和IL-1β的转录(71)。因此,亚甲基丁二酸是KEAP1的内源性调节剂,在炎症的控制过程中起到“关闭开关”的作用。此外,通过细胞可渗透的亚甲基丁二酸衍生物4-octylitaconate或SFN激活NRF2,通过降低mRNA稳定性来抑制干扰素基因(STING)的衔接子分子刺激物的表达,从而抑制I型干扰素(IFN)的产生(73,74)。
NRF2-KEAP1信号通路和非肿瘤性疾病
在细胞和动物模型中已经发现了大量关于NRF2对多种慢性疾病保护作用的信息,许多研究使用了NRF2敲除小鼠(75)。关于人类,最近的一项研究为使用系统医学方法分析NRF2对抗慢性病奠定了基础,并且基于NRF2与其他分子(NRF2相互作用组)的连通性以及经验证据,建立了第一个NRF2相关疾病(NRF2病变)的图谱(9)。NFE2L2的功能性遗传变异与疾病风险之间的关联研究不断提供有关NRF2在确定人类对慢性病易感性中作用的令人信服的证据。此外,这些遗传学研究支持这样一种假设,即对于NFE2L2的某些功能变化,模仿NRF2表达的微小变化的药物可能具有治疗价值。下面,我们将简要描述NRF2参与人类慢性疾病的一些里程碑性的成果,并重点介绍现有的遗传学证据。
自身免疫性疾病
氧化组织损伤和细胞凋亡导致半抗原或受损大分子的形成,增加了自身免疫反应的风险。由于NRF2调节的酶在许多化学物质的解毒中起着关键作用,因此它提供了一种保护机制,防止对自身免疫发病机制的环境易感性(79)。此外,NRF2抑制促炎T辅助细胞1(TH1)和TH17细胞反应并激活免疫抑制调节性T细胞(Treg)和TH2细胞(80)。NRF2在自身免疫中的相关性已经在实验性自身免疫性脑脊髓炎(EAE)中进行了广泛研究,其是MS的小鼠模型,MS是一种慢性炎性疾病,其特征在于自身反应性免疫细胞浸润到中枢神经系统中。在EAE小鼠模型中,NRF2的缺乏加剧了疾病(81),而KEAP1的敲低(52)或用一系列NRF2激活剂(包括氰基三萜类化合物和SFN)(82,83)治疗减弱了其严重性。在人类中,通过IFNβ治疗患者基因表达谱确定NRF2是长期抗氧化反应和神经元保护的潜在介质(84)。如下所述,一些生物制药公司正在利用强有力的证据表明NRF2的活化在MS中具有治疗价值。事实上,迄今为止唯一获得美国食品药品管理局(FDA)和欧洲药品管理局(EMA)DMF批准的NRF2激活剂正在由Biogen销售,用于治疗缓解复发型MS和银屑病(85)。NRF2的保护作用已被建议用于其他自身免疫性疾病,如系统性红斑狼疮(86, 87)、干燥综合征(86)、类风湿性关节炎(88, 89)、白癜风(90-92)和STING依赖性干扰素病(74)。
呼吸系统疾病
肺通过释放促炎细胞因子和趋化因子对包括烟雾在内的各种环境毒物作出反应。这一主要效应通过循环单核细胞、中性粒细胞和T细胞的募集产生渐进的低度炎症反应,这些细胞释放额外的炎症介质、ROS和金属蛋白酶。最终结果是肺实质受损的恶性循环,导致慢性阻塞性肺病(COPD)或肺气肿(93)。NRF2减轻由ROS和炎症引起的肺实质负担。香烟烟雾是COPD和肺气肿发展的主要危险因素。
NFE2L2-/-小鼠对香烟烟雾引起的肺气肿的发展比野生型更敏感(94)。此外,长期暴露于香烟烟雾后肺气肿的发展与细胞保护基因的表达下降有关(95),而五环氰基酮CDDO-咪唑啉保护NFE2L2+/+小鼠对抗香烟烟雾诱导的氧化应激、肺泡细胞凋亡、肺泡破坏和肺动脉高压,而不是NFE2L2-/-小鼠(96)。NRF2的遗传上调(通过Clara细胞中的Keap1缺失,其在鼠上呼吸道中是丰富的)具有类似的保护作用(97)。
在人类中,与没有肺气肿的患者相比,吸烟相关肺气肿患者的肺泡巨噬细胞中NRF2转录信号降低(98)。人类NFE2L2基因启动子包含一个含有三个单核苷酸多态性(SNP)序列的单倍型和一个调控NRF2表达的三重重复多态性。具有低至中等启动子活性的COPD患者更容易发生呼吸衰竭(99)。SFN对NRF2的药理学激活,或一种干扰KEAP1与NRF2相互作用的化合物,逆转培养肺泡巨噬细胞和从COPD患者中分离的单核细胞来源巨噬细胞受损的细菌吞噬作用(70,100)。然而,每日口服一定剂量SFN(西兰花芽苗提取物)的慢性阻塞性肺病患者并没有导致NRF2依赖性基因表达或肺泡巨噬细胞和支气管上皮细胞炎症标志物的一致性变化(101),说明将细胞培养研究结果转化为人类结果的一些挑战。其他慢性肺病,如特发性肺纤维化、慢性结节病和过敏性肺炎,被发现在支气管肺泡灌洗液中表现出NRF2表达增加和内源性抗氧化剂水平增加,表明对病理性ROS水平的适应性反应不成功(102)。
胃肠疾病
胃肠道经常受到外来生物的挑战,这可能导致形成病理水平的ROS,并可能引发IBD(103)。胃肠道也是膳食及其合成类似物在药理学上激活NRF2的重要部位之一(23, 104-106)。在小鼠中,右旋糖酐硫酸钠(DSS)诱导的实验性结肠炎在NRF2缺陷小鼠中引起的IBD要比野生型小鼠严重(107)。在人类中,NRF2在肠细胞对炎症应激适应的相关性在IBD患者的一项全面转录组研究中得到证实,证明了NRF2在减弱肠道巨噬细胞应激反应中的作用(108)。重要的是,在一组日本患者中发现了NFE2L2基因中特定SNP与溃疡性结肠炎发病风险之间的遗传关联(109)。NFE2L2启动子中3个SNP功能单倍体NRF2表达略有降低,与胃黏膜炎症和幽门螺杆菌感染相关消化性溃疡的发生发展有关(60)。
肝脏是抵御食物、外来生物制剂和口服活性药物的第一道防线,因为从消化道流出的大部分血液通过肝门静脉直接流到肝脏。早期使用NFE2L2-/-小鼠证实了NRF2对对乙酰氨基酚的保肝作用(110),NRF2的遗传活化也是如此(111)。正如可能预期的那样,喂食酒精(4-7天)的NFE2L2-/-小鼠比同样喂养的野生型小鼠明显更容易脂肪肝加重、肝脏炎症反应和肝功能衰竭,这在一定程度上可能是SREBP脂源性转录因子的激活和对乙醛解毒的相对无能(112)。通过观察NRF2在小鼠肝部分切除后对肝脏再生的阻碍,发现NRF2调控NOTCH1基因表达(113),这一机制也参与了NRF2介导的造血干细胞祖细胞功能改善和电离辐射后骨髓抑制(114)。在遗传性血色素沉着症伴铁超载的小鼠模型中(由于稳态铁调节基因突变),敲除NRF2增加了小鼠肝脏内导致肝纤维化的坏死炎症病变(115)。与这些观察结果一致,原发性胆管炎和肝硬化患者NRF2表达减少(116。)
代谢性疾病
代谢性疾病又称代谢综合征,以腹部肥胖、高血压、高甘油三酯血症、低水平高密度脂蛋白和空腹高血糖为特征。它与2型糖尿病(T2DM)和相关的合并症如血管功能障碍、心血管疾病、肾小球肾炎、非酒精性脂肪性肝炎(NASH;见下文)和认知障碍密切相关。尽管T2DM及其合并症的特征在于线粒体产生过量超氧化物(117),但NRF2缺失小鼠不会自发地发展成糖尿病。实际上,敲除小鼠表现出增加的胰岛素敏感性,这归因于ROS介导的蛋白酪氨酸激酶磷酸酶1B的抑制,该蛋白拮抗胰岛素信号(118)。相比之下,db/db背景下小鼠中KEAP1敲除或敲低对NRF2的遗传激活,或db/db小鼠中NRF2的药理活化,抑制了T2DM的发生(119)。因此,似乎当胰腺β细胞代谢挑战合成胰岛素以应对高血糖时,所产生的ROS负担可能会导致氧化损伤、损害反应、导致T2DM(120,121)。如果这个结论是正确的,那么很可能在饮食诱导的T2DM小鼠中,TBE-31或SFN激活NRF2后,胰腺功能的改善对葡萄糖处理的改善有显著的作用(122,123)以及富含SFN西兰花芽提取物对T2DM失调肥胖患者空腹血糖和糖化血红蛋白(HbA1c)的降低作用(123)。除了增加对胰腺β细胞的抗氧化应激能力之外,NRF2活化可通过增加糖原分支酶和磷酸化酶b激酶α的表达来抑制骨骼肌中的糖原分解,从而有助于改善全身葡萄糖稳态(119)。在糖尿病肾病小鼠模型中,SFN或肉桂醛对NRF2的药理激活已被证明可抑制氧化应激以及转化生长因子β1信号传导和纤维化(124)。
NRF2可能在胰岛素抵抗中发挥关键作用,因为它在糖尿病患者的单核细胞中表达很低(125)。令人信服的证据来自遗传关联研究,表明导致低NRF2表达的一些NFE2L2多态性导致病理性ROS和T2DM的风险增加(126-128)。关于并发症,由于KEAP1启动子中CpG岛的去甲基化导致的抗氧化保护失效与糖尿病白内障有关(129)。
NRF2也可能对NASH的发展具有保护作用。在使用缺乏蛋氨酸和胆碱饮食的NASH小鼠模型中,NRF2缺失小鼠比野生型小鼠死于该疾病的速度要快得多(130,131)。在更为生理的NASH模型中,喂食高脂肪饮食的NRF2缺失小鼠也会出现肝脏脂肪变性和炎症恶化,这表明需要NRF2来抑制这些症状的发生(118, 132)。此外,野生型而非NRF2敲除小鼠,在长期高脂肪、高果糖饮食使其变得肥胖和胰岛素抵抗后,通过三环氰基酮TBE-31对NRF2药理激活后,逆转了胰岛素抵抗,抑制肝脏脂肪变性,改善NASH和肝纤维化(122)。TBE-31对NRF2的药理激活拮抗SREBP1c和ChREBP及其脂质生物合成靶基因的表达,这可能涉及AMPK的激活(122, 133)。此外,在小鼠肝脏中,NRF2的基因激活可拮抗糖异生酶PEPCK和G6PC的表达,这可能也涉及AMPK(133)。在NASH患者的肝脏活检标本中,发现病理性ROS水平升高、NRF2基因表达增加,提示其试图减轻氧化和炎症(134)。
心血管疾病
ROS的产生和处理之间的不平衡导致破坏性氧化物质的过度形成,已被认为在许多心血管疾病中发挥作用,如高血压、动脉粥样硬化、糖尿病血管疾病、心肌缺血再灌注损伤和心力衰竭(135)。此外,血管壁内的低度持续性炎症是动脉粥样硬化易损斑块的形成过程和发展特点,所以动脉粥样硬化斑块易破裂,进而导致血栓形成和终末器官缺血。鉴于NRF2是抗氧化防御的关键调控因子,且具有直接的抗炎特性,因此可以合理地假设NRF2可以预防与ROS相关的心血管疾病。事实上,包括富马酸盐和硫化氢在内的NRF2激活剂在许多心血管疾病动物模型中显示出了保护作用,如心脏缺血再灌注损伤(136, 137)和心力衰竭(138, 139)。NRF2在高胆固醇血症小鼠模型动脉粥样硬化中的作用就不那么直接了:在骨髓来源细胞中NRF2的缺失加重了动脉粥样硬化(140, 141),而根据病变发展程度评估的NRF2全面缺失减轻了动脉粥样硬化(142-145),但增加了斑块炎症和易损性(145)。然而,一些NRF2激活剂已被证明在多种动脉粥样硬化动物模型中具有动脉粥样硬化保护作用(146-148)。由收缩性、细胞骨架蛋白和分子伴侣蛋白突变引起的心脏疾病约占遗传性心肌疾病的30%(149)。在某些情况下,还原而不是氧化应激,再加上蛋白质聚集,与心肌病有关。血液透析患者全身炎症和病理ROS的形成与NRF2的下调有关(150),位于NFE2L2启动子的多态性与这些患者死亡率的增加有关(151)。NRF2信号通路的破坏可防止还原性应激并延缓过量表达心脏中CRYAB(α-结晶B链)小鼠蛋白质性心脏病(152)。因此,正如这篇综述所强调的,针对NRF2的环境非常重要——越多不一定越好。
神经退行性疾病
在各种形式的神经退行性疾病中,NRF2和蛋白稳定之间的联系尤其密切,因为这些疾病的特点是蛋白异常聚集(43)。NRF2在蛋白质病中具有保护作用的令人信服的证据已经在阿尔茨海默病(AD)模型的淀粉样病变(153,154)、tau病变(155)或两者皆有(40, 156, 157)以及帕金森病(PD)模型的α-突触核蛋白病变(158-160)中得到证实。在人类,APP受损和tau损伤神经元NRF2及其靶蛋白p62 / SQSTM1表达增加(40,161)。这些发现与最近报道的NRF2上调巨自噬(40)和分子伴侣介导自噬(39)的基因表达作用是一致的,这是清除APP、tau和α-突触核蛋白的两种必要机制。NRF2上调保护神经元免受突变型α-突触核蛋白和leucinerich重复激酶2(LRRK2)的毒性(162),LRRK2也导致与错误折叠蛋白质聚集相关的神经变性(163)。奇怪的是,尽管NRF2激活增加了α-突触核蛋白的降解,但错误折叠的弥漫性LRRK2被隔离到包涵体中。
NRF2在星形胶质细胞和小胶质细胞中的作用尤其重要,因为它们在大脑中表达水平最高(图2b)。通过使用带有ARE驱动的碱性磷酸酶报告基因的转基因小鼠,我们发现星形胶质细胞在几种神经变性模型中对NRF2的激活反应高度敏感(164),这表明NRF2通过对即将死亡的神经元提供代谢和GSH前体而在星形胶质细胞的培育作用中具有相关性(46, 165)。小胶质细胞作为大脑的免疫应答细胞,在NRF2介导的应答中也起着至关重要的作用(166, 167)。在帕金森毒素甲基-4-苯基-1,2,3,6-四氢吡啶致小鼠PD模型中,NRF2可调节小胶质细胞动力学,从而降低COX2、NOS2、IL-6和TNF的生成,并增加多种抗炎标志物的水平(55, 168)。tau损伤神经元激活小胶质细胞似乎至少部分地通过与分形趋化因子相互作用有关。在小鼠和人类,tau损伤神经元释放出分形趋化因子,抑制邻近的小胶质细胞中GSK3并导致NRF2蛋白的增加。反过来,NRF2减少TNF和IL-6的释放,并参与小胶质细胞的重编程,以达到愈合伤口(161)。
一些NRF2靶基因,包括HMOX1、NQO1、GCLM和SQSTM1,在AD和PD脑中上调(159,169-171)。通过对NFE2L2启动子中3个SNP的功能单倍型与疾病风险之间关系的遗传分析,证明了这种上调的相关性。在肌萎缩性脊髓侧索硬化症(ALS)中,一种保护性的单倍体等位基因与疾病发作延迟4年有关(172),但另一项研究没有发现明显的关联(173)。在AD中,另一个单倍型变异与早2年发病有关(76)。已经报道了PD的更详细证据。最初,保护性单倍型与瑞典队列中疾病发作延迟相关,并且波兰队列PD患者的风险降低(77)。之后,这些研究结果在四个独立的欧洲病例对照研究中得到了重复(174),但台湾人口却没有(175),这表明存在种族和/或环境差异。NRF2的药理激活也有望用于治疗亨廷顿病(Huntington disease, HD)和弗里德雷希共济失调(Friedreich ataxia, FRDA),对于这两种疾病来说,氧化应激和炎症是重要的病理驱动因素。在小鼠、来自HD和FRDA患者的模型生物体和培养细胞中进行的研究报告了NRF2信号通路受损(53,176 - 178)以及药理学NRF2激活剂的有益作用。因此,三唑衍生物对NRF2的激活抑制了HD原代小鼠小胶质细胞和星形胶质细胞以及来自HD患者的培养单核细胞促炎细胞因子的释放(53),并且在离体HD大鼠皮质纹状体脑切片和HD果蝇模型中具有神经保护作用(179)。在HD、DMF和三萜类化合物的小鼠模型中,CDDO-乙基酰胺和CDDO-三氟乙基酰胺改善了动物的脑病理和行为(180,181)。用SFN和环状氰基酮TBE-31或RTA-408处理改善了FRDA小鼠模型成纤维细胞、小脑颗粒神经元以及FRDA患者成纤维细胞的线粒体功能和抗氧化能力(182,183)。总而言之,现有证据表明NRF2的适度激活对大脑具有神经保护作用。
NRF2-KEAP1信号通路和癌症
据报道,NRF2具有抗肿瘤和促肿瘤作用。在这里,我们简要总结了目前对NRF2在癌症中作用的理解,并参考了最近在癌症背景下讨论NRF2的全面综述(12)。
在非恶性细胞中,NRF2激活对氧化剂诱导的遗传损伤以及化学和物理致癌物具有抵抗性,这是由于NRF2激活增强了机体的防御能力,包括抗氧化(184)、放射性保护(114)以及加速DNA损伤剂的生物转化和清除(185)。此外,NRF2转录反应的激活似乎将ROS水平维持在低于对肿瘤发生关键蛋白质发送信号所必需的水平,例如磷脂酰肌醇-4,5-二磷酸3-激酶(PI3K)、丝裂原活化激酶、缺氧诱导因子1和NF-κB(186)。
相反,在肿瘤发展的早期阶段,选择具有NRF2组成型激活的癌细胞作为能够适应恶劣微环境、化疗、放疗或高内源ROS水平的方式。此外,在快速增殖的细胞中,NRF2通过增强核苷酸(26)和氨基酸(187)的生物合成来支持中间代谢,但也会产生代谢失衡和对谷氨酰胺酶的依赖,这可能用于治疗(188,189)。
KEAP1的体细胞功能缺失突变或NFE2L2的功能获得突变在非小细胞肺癌和其他一些环境因素是重要病因的癌症中很常见(190)。癌症基因组图谱(TCGA)数据库中的NRF2突变综合目录在10364例中识别出226个独特的NRF2突变肿瘤,其中33种肿瘤类型中有21种发生了功能获得性NRF2突变(191)。这种突变的频繁发生表明NRF2抑制剂在癌症治疗中的潜在益处,从而引发对此类药物的研究(Box 1)。这些NRF2抑制剂代表了药理学的一个完全开放的领域,目前处于概念验证研究的水平。NRF2抑制的替代方法包括靶向依赖NRF2和/或在KEAP1突变癌细胞中选择性表达的蛋白。例如谷氨酰胺酶(188, 189)和NR0B1的抑制剂,NR0B1是一种非典型的孤儿核受体,在多聚体蛋白复合物中调节KEAP1突变细胞的转录(192)。然而,值得注意的是,目前尚无证据表明单单KEAP1或NFE2L2突变能够引发恶性转化,与致癌驱动因子共发生似乎是必要的,PI3K通路改变与恶性转化的相关性最强(193)。
最能说明问题的是,KEAP1亚等位基因敲低的NRF2全基因组上调小鼠不会自发形成肿瘤(194, 195),而小鼠肺中同时缺失Keap1和PTEN则会促进腺癌的形成(195)。重要的是,由此产生的肿瘤具有免疫抑制微环境的特征,可通过免疫检查点抑制剂进行治疗(195)。很明显,NRF2基因激活是肿瘤发生的重要因素,因为癌基因(KRASG12D)激活啮齿动物NRF2信号可以增加肿瘤发展(196),而KEAP1基因敲低(也导致NRF2信号激活)强烈阻碍NOTCH1驱动的肿瘤发生(197)以及紫外线辐射介导的皮肤癌发生(51, 198)。值得注意的是,在后一种模型中,同时丧失NRF2可以消除KEAP1敲低对皮肤癌发生的保护作用(199)。 NRF2的这种两面作用得到广泛的实验证据支持,在这些实验证据中,NRF2的激活增强了抗肿瘤免疫,以防止肺癌的发生,但只有在肿瘤发生后才会加速恶性生长(200)。此外,与野生型同窝小鼠相比,接受化学诱导致癌作用的NFE2L2敲除小鼠表现出肿瘤数量增加(启动增强)但体积小得多(进展受损)(201)。因此,低NRF2活性促进癌发生的起始,而持久的组成型高NRF2活性可以驱动癌症进展和对治疗的抗性(202)。
NRF2调节剂的临床开发
富马酸酯。富马酸酯代表一组NRF2激活剂,工业上对此进行了大量的研究。临床最成功的例子是DMF(化合物1,图3a),它于1994年被批准用于治疗银屑病,并且基于其在MS的EAE小鼠模型中的功效(203),于2014年被Biogen(Tecfidera)重新用于治疗复发-缓解型MS。在发现NRF2之前,对啮齿动物的早期研究报道DMF是NRF2转录靶点GST和NQO1的强效诱导剂(204),后来发现DMF代谢物富马酸单甲酯(MMF;化合物2,图3a)与KEAP1中的C151反应,激活NRF2(203)。自从发现DMF的诱导剂活性以来,安全性一直是一个优先考虑的问题,并注意到获得显著酶诱导剂所需DMF浓度对啮齿动物具有良好的耐受性(204)。DMF在两个III期试验中表现出良好的安全性和耐受性(205, 206),自其商业化以来,它已成为近年来最成功的新药之一。然而,Tecfidera的一个缺点是出现轻度至中度腹痛、潮红、腹泻和恶心。尽管可以控制这些不良反应,但是更严重的症状是在治疗开始时白细胞计数低的患者发生白细胞减少症。事实上,用DMF治疗的动物神经系统中粒细胞的水平远低于未接受该药物治疗的动物(207)。该作用与NRF2无关,但很可能是由于羟基羧酸受体的活化,因为缺乏该蛋白质的小鼠具有高水平的粒细胞,无论它们是否用DMF处理(207)。这些观察结果强调了仔细选择治疗纳入标准的重要性。
如上所述,DMF在体内代谢为MMF,通过在C151处形成加合物使KEAP1失活(203)。由于这种代谢转化,一些生物制药公司正在开发具有缓慢和持续释放MMF的化合物,与DMF相比,其显示出改善的生物利用度和更少的副作用(表1)。Alkermes(被Biogen收购)开发了富马酸迪罗西酯(BIIB098,以前为ALK8700,化合物3;图3a),一种具有降低GI副作用的MMF前药,现在正在进行MS的III期临床试验(NCT03093324)。 XenoPort(由Arbor Pharmaceuticals收购)开发了富马酸富马酰胺,一种MMF前药(XP23829,化合物4,图3a)。与DMF相比,XP23829在临床前模型中具有更高的溶解度和渗透性,口服给药后更大的吸收,改善功效和降低GI副作用,并且现在处于斑块状银屑病的II期临床试验中(NCT02173301)。Catabasis制药公司正在开发另一种MMF和二十二碳六烯酸(CAT4001)的化学连接偶联物,与单独的生物活性分子相比,这种偶联物具有增强细胞靶向性、有效性、安全性和耐受性的潜力。特定的酶释放细胞内的两个组件,同时调整多个生物目标,包括细胞和动物模型中的NRF2和NF-κB, 并显示出治疗神经退行性疾病(如FRDA和ALS)的前景。V ClinBio正在开发一种类似的技术,用于治疗MS和银屑病的MMF和二十碳五烯酸(VCB101,化合物5,图3a和VCB102)的结合物(表1)。
萝卜硫素(SFN)。SFN一种广泛应用的天然亲电NRF2激活剂 (化合物6,图3a)。该化合物最初由Paul Talalay和Yuesheng Zhang作为十字花科植物提取物NRF2靶酶NQO1的主要诱导物分离得到的(208),并与KEAP1的C151相互作用(14,209)。值得注意的是,SFN可通过血脑屏障(55),并在许多神经系统疾病的临床前模型中具有保护作用(210)。在一项针对患有自闭症谱系障碍年轻男性的双盲安慰剂对照临床试验中,口服富含SFN的西兰花芽提取物胶囊,通过异常行为检查表和社会反应量表评估,在行为方面改善显著;这些量表上的总分数在停止治疗后逆转至治疗前水平(211)。三日龄的西兰花芽富含葡萄糖苷,一种SFN前体分子,可被植物黑芥子酶水解,释放出SFN和葡萄糖(212)。类似的β-硫葡糖苷酶存在于肠道微生物群中;因此,释放的SFN实际水平高度依赖于饮食习惯和微生物组成,受抗生素治疗的影响(213)并表现出昼夜节律(214)。并且,无论是芽苗提取物还是高度纯化的SFN,已有超过500名受试者接受超过25,000剂量,证明了具有高度安全性。目前有超过20个正在进行的临床试验。
考虑到天然化合物知识产权的相关问题、SFN在室温下不稳定以及需要精确控制剂量,Evgen Pharma开发了SFN的药物形式,SFX-01(化合物7,图3a) 。 SFX-01是化学合成的SFN,包裹在环糊精中,形成稳定的固体形式的丸剂或胶囊,具有良好的生物利用度。SFX-01目前正在用于蛛网膜下腔出血(NCT02614742)和转移性乳腺癌(NCT02970682)临床试验(表1)。Evgen还有一系列基于SFN的新型类似物,这些类似物正处于临床前评估阶段。此外,许多营养保健品公司正在生产含有NRF2诱导剂的制剂,包括SFN,具有不同程度的标准化。其中一个例子是来自Nutrinov实验室的Prostaphane,它是一种稳定的游离SFN,从花椰菜种子中提取,在前列腺癌的安慰剂对照临床试验中显示了在根治性前列腺切除术后控制生化复发的优势(215)。SFX-01和Prostaphane的生物利用度和效力等同于稳定性较差的SFN(216)。另一个例子是胶囊膳食补充剂Avmacol(Nutramax Laboratories),含有来自精细研磨的花椰菜种子的萝卜硫苷和冷冻干燥的西兰花芽粉末以提供黑芥子酶。该补充剂目前正在临床试验中用于调节与自闭症谱系障碍、精神分裂症和环境污染有关的疾病症状和/或生物标志物(217)。
氰基酮三萜。越来越多的证据表明,富马酸酯、SFN以及与KEAP1共价结合的其他小亲电试剂可以从更大的支架结构中受益,以赋予更好的选择性(和可能的效力)和更可控的药代动力学和/或药效学特性。含五环迈克尔受体的氰基酮三萜类化合物的开发可满足这些要求。这些五环三萜类化合物最初由Michael Sporn、Gordon Gribble和Tadashi Honda开发,来自天然产物齐墩果酸(218, 219),这些五环三萜类化合物是迄今已知最有效的亲电子NRF2激活剂(220),由KEAP1中的C151检测,目前正在由Reata Pharmaceuticals和Kyowa Hakko Kirin进行临床开发。
两种临床上先进的化合物,bardoxolone methyl(BARD;化合物8,图3a)和omaveloxolone(化合物9),能使代谢正常化、增加线粒体能量产生、增强细胞抗氧化能力和降低ROS水平(221)(表1)。在临床前研究中,BARD或类似物已被证明可通过减少炎症、纤维化和氧化应激以及增加肾小球的过滤表面积来改善肾功能(222, 223)。通过修改选择标准以排除有早期心力衰竭迹象的患者(B型利钠肽升高),BARD目前正在临床试验中测试几种与慢性肾病(CKD)相关的罕见病症,大多数没有经批准的治疗方法,包括Alport综合征(NCT03019185)、常染色体显性遗传性多囊肾病、免疫球蛋白A(IgA)肾病、1型糖尿病和局灶性节段性肾小球硬化(NCT03366337),其中促炎和促纤维化过程造成肾小球硬化和肾功能受损(224)。BARD治疗有可能延缓或预防肾小球滤过率下降,而肾小球滤过率下降会导致Alport综合征和其他罕见形式CKD患者需要透析或移植。因此,在患有T2DM和CKD患者的II期临床试验中(NCT02316821),BARD治疗可使直接测量肾小球滤过率的统计学显著增加,并且在排除具有液体潴留风险患者后,耐受性良好。在这些结果的基础上,Kyowa Hakko Kirin在患有G3期或G4期CKD的糖尿病患者中,在日本SAKIGAKE指定系统的支持下,开展了III期临床试验(NCT03550443)。
此外,BARD正在用于研究肺动脉高压(PAH)(NCT03068130)和结缔组织病(CTD-PAH)引起的PAH(NCT02657356),这是一种导致心力衰竭和死亡的严重和进行性疾病。CTD-PAH与特发性病因之间的差异很大程度上归因于炎症、自身免疫和系统性血管病变之间复杂的相互作用,这些相互作用促成了结缔组织疾病的发病机制。由于NF-κB的上调,CTD-PAH患者的炎症加重(225),对现有血管扩张剂治疗的反应性低于特发性PAH患者,预后较差(226)。在II期研究中,对CTD-PAH患者进行了BARD测试,目前正在III期研究中进行测试。Omaveloxolone目前正在FRDA患者(NCT02255435)的II期临床试验中进行测试,这是一种遗传性神经肌肉疾病,其NRF2被下调(176-178)。值得注意的是,目前尚无批准治疗FRDA的疗法(227)(表1)。
硝基脂肪酸。脂肪酸的硝化衍生物(NO2-FAs)是内源性信号介质,在代谢和炎症疾病的临床前动物模型中具有抗炎和抗纤维化活性(228)。硝基烯烃基团赋予其β-碳亲电性,促进与亲核试剂如半胱氨酸的可逆NO2-FA迈克尔加合物的快速形成,这种改性称为硝基烷基化(22, 230)。已经表明,硝基油酸(NO2-OA; 9-硝基十八碳-9-烯酸)与KEAP1中的半胱氨酸反应,包括C273和C288,从而激活NRF2(231,232)。该反应的可逆性阻止了稳定的NO2-FA硫醇加合物积累的可能性,这可能导致细胞毒性(219,230,233)。实际上,I期安全性评估证明了先导化合物CXA10(10-NO2-OA; 10-硝基-十八碳-9-烯酸,NO2-OA的特异性区域异构体)(化合物10,图3a)在药理活性剂量下对人体是安全的(234)。目前,Complexa正在开发CXA10作为局灶性节段性肾小球硬化和PAH的治疗方法(234)(表1)。最近报道了来自α-生育酚的硝基烯烃合成和生物学评价(235)。
羟胺。一个特殊的挑战是提供可以通过血脑屏障的NRF2激活剂。给药途径(注射、口服或局部)对特定疾病也至关重要。Othera Pharmaceuticals(现由Colby Pharmaceutical收购)正在开发一种具有抗氧化特性的二取代羟胺(OT551),局部应用,通过靶向KEAP1抑制氧化应激和疾病相关炎症(化合物11,图3a)。在临床前研究中,该化合物的眼用溶液保护视网膜色素上皮细胞和光感受器免受氧化损伤和炎症反应,并且一项关于年龄相关性黄斑变性的II期试验已证明可有效预防视力丧失的进展(NCT00485394)(表1)。
TFM735。一种高通量筛选报告子策略评估了包含融合到LacZ (NRF2d-LacZ)236的NRF2氨基末端区域嵌合蛋白的稳定性,该策略确定TFM735为先导化合物(化合物12,图3a)。TFM735以C151依赖性方式激活NRF2,并抑制受刺激的人外周血单核细胞中IL-6和IL-17合成以及小鼠EAE进展(237)。该化合物目前由Mochida Pharmaceuticals进行治疗MS的临床前开发(表1)。
NRF2-KEAP1蛋白-蛋白相互作用抑制剂。近年来,非亲电性非共价化合物的发展受到关注,这些化合物可以直接干扰KEAP1和NRF2之间的PPI(238),或者干扰KEAP1和CUL3之间的PPI(239)。通过直接干扰KEAP1和NRF2或CUL3之间的相互作用,化合物的作用方式不需要共价结合组分。该方法具有潜在的优点,包括探索NRF2诱导物的新化学型的范围,由于与KEAP1的不同相互作用模式导致的不同药效学以及由于半胱氨酸非依赖性结合机制导致的不同脱靶效应谱。有证据表明,亲电子和非亲电子(PPI)KEAP1抑制剂的生物学效应谱不同,一个例子是非亲电试剂诱导线粒体自噬的能力,与SFN和DMF等亲电试剂相比;这表明两种化合物类别之间药理活性和治疗效用的潜在差异。
PPI抑制剂的设计是以KEAP1的Kelch结构域的晶体结构可用性为指导的(241-243)(图3b)。已经报道了几种类型的PPI抑制剂;根据制药行业投入开发的主要化学类别如图3b所示。萘双磺酰胺化合物起源于Biogen的高通量筛选(244),随后由中国药科大学Jiang等人提供化合物13(CPUY192018)和类似物(245)。该双羧酸化合物是KEAP1的高亲和力配体,在低微摩尔浓度下诱导ARE依赖性基因的表达。Astex和葛兰素史克还从基于X射线晶体学碎片的筛选程序中鉴定了含磺酰胺的铅化合物。在这种情况下先导化合物的例子是化合物14,KEAP1-NRF2 PPI低纳摩尔抑制剂,它能增加COPD患者上皮细胞中NRF2靶基因NQO1的表达。该化合物能够在静脉内给药后减轻大鼠的肺部炎症,并有效抑制臭氧诱导的支气管肺泡液中白细胞的积累并恢复GSH浓度(246)。来自该系列的化合物已经在几种模拟肺病特征的氧化应激动物模型中显示出功效,并且目前正在开发用于临床评估。已经开发了另外两类不是磺酰胺的单酸性PPI抑制剂。第一类是由胡等人在Rutgers开发(247),由Evotec和UCB Pharma详细描述(248),包括四氢异喹啉(例如化合物15),它们是KEAP1-NRF2 PPI的低微摩尔抑制剂。第二类是由Toray Industries和RIKEN开发的,并加入了恶二唑基序;化合物16是PPI抑制剂,具有微摩尔范围内的结合活性(249)。突出显示的化合物或其类似物已与KEAP1 Kelch域共结晶;图3b显示了化合物13-16所获得的结合位点亚袋的占用情况,并为这些配体未来的精炼提供了思路。
C4X Discovery利用计算化学、配体核磁共振光谱和蛋白质结晶学方法以及Keapstone利用基于结构的药物设计方法联合开发了新的PPI抑制剂。学术团体也在为非亲电NRF2诱导物研究做出贡献(250,251)。
如何使PPI抑制剂具有适当的药物代谢和药代动力学特性,用于外周神经系统和中枢神经系统,仍然是一个重大挑战,后者是一个需要具有强安全性NRF2激活剂用于长期给药等条件的领域,如AD和PD。此时,非亲电性PPI抑制剂改善靶向选择性的潜在优势被当前化合物相对较高的分子质量、阻断较大KEAP1-NRF2界面的要求以及极性官能团赋予KEAP1紧密结合亲和力的需要所抵消。因此,目前通过体外研究鉴定的许多原型化合物表现出较差的吸收、分布、代谢和排泄性质。例如,萘双磺酰胺(化合物13)对DSS诱导的小鼠结肠炎具有保护作用,在低微摩尔范围内激活NCM460培养细胞中NQO1表达,并在体外与纳米级Kd结合,说明蛋白质结合与生物活性之间的相互抵消(252)。
大多数PPI抑制剂的体内评价都集中在外周炎症。例如,化合物NK-252(化合物16的类似物)被Toray Industries鉴定为相当弱的KEAP1抑制剂,但对H2O2诱导的细胞毒性具有保护作用并且降低了胆碱缺乏的l-氨基甲酸盐饮食大鼠(NASH模型系统)的纤维化评分(253)。
用KEAP1非依赖性药物靶向NRF2。越来越多的证据表明,NRF2除了KEAP1所发挥的作用外还表现出多层调节,如转录(196)、表观遗传(254,255)、共价蛋白修饰(258,257),以KEAP1非依赖性方式进行蛋白酶体降解(258-261)以及NRF2二聚体伴侣的靶向结合ARE序列的调节(262,263)。在药理学发展水平上,ARE介导的基因调控的KEAP1非依赖性机制涉及调节转录抑制因子BACH1(广泛复合物,tramtrack、bricàbrac和cap'n'collar同源体1),该机制抑制NRF2调控基因亚基的失活,尤其是HMOX1(28)。vTv 疗法现在正在分析该亚基是否足以引起治疗用处。他们开发了一类新的非亲电子小分子,可以独立于KEAP1抑制BACH1与某些ARE驱动基因的结合(264)。
挑战与思考
药效学评价。考虑到NRF2的半衰期很短,即使在药物诱导稳定后,也需要通过间接指标来推断其在病变器官中的激活情况,从而确定最合适的治疗方案。一种可能性是分析可接触的细胞或组织(如外周血单核细胞、鼻腔灌洗液细胞、脱落的膀胱细胞、颊细胞和皮肤)中的药物分布和NRF2基因表达特征,希望NRF2的转录组特征反映了其他组织(如肺,肝和脑)的局部参与(265-267)。群体不可避免地暴露于有害环境化学物质的情况下,尿液中相应代谢物的水平已被用作NRF2介导的药效学作用生物标志物(268,269)。
NRF2通路的激活表现为依赖于时间、组织、剂量、发病时间和持续时间的差异基因表达。表2显示了NRF2调控基因产物代表性亚群的半衰期。由于其中许多蛋白质的半衰期相当长,其生物化学活性的持续时间预计将远远长于NRF2稳定的时间间隔或其药理激活剂的存在,后者通常在数小时内被清除。因此,NRF2活化分子的药效学观察时间比NRF2水平要长得多,且与血浆药物浓度不一致。因此,NRF2激活剂TBE-31(一种与上述三萜类化合物密切相关的三环氰基酮)在小鼠皮肤和血浆中的半衰期为10小时(51,270),而NRF2依赖的NQO 1诱导蛋白在最后一剂局部应用药物3天后仍可在皮肤中检测到(51)。因为NRF2激活剂导致“刺激产生或抑制控制测量效果的因素”,所以“间接药效学反应”模型可能是用于测试NRF2激活剂药效学走向临床应用的更合理方法(272,272)。
NRF2活化药物在相关组织中的浓度和分布,以及它们的溶解度、细胞通透性、代谢稳定性和蛋白结合,也将在NRF2活化程度中发挥重要作用,最终达到预期的长效药效学效果。因此,优化临床有意义的药物暴露需要通过一个假定的NRF2激活剂分析剂量依赖性基因调控。此外,NRF2靶向参与研究必须考虑患者的年龄和表现状态,因为在年龄较大和健康状况较差的受试者中,药物激活NRF2或促进“适应性反应”的能力似乎有所下降。最近在DMF复发缓解型和多发性硬化症患者的临床试验中发现,NRF2转录靶NQO1的诱导程度与患者年龄呈负相关, 在4 - 6周高NQO1蛋白含量的患者在开始治疗后的第二年更有可能没有疾病活动的证据(267)。
药物选择性。KEAP1中高活性半胱氨酸的鉴定对于理解亲电化合物与泛素连接酶底物适配器共价作用的机制至关重要。结构信息表明,某些作用于C151的药物可能会干扰与CUL3-RBX1的相互作用(273, 274),从而使KEAP1库被结合的NRF2饱和,并允许新合成的NRF2逃避降解: 这是用亲电试剂n -碘乙酰- n -生物素己二胺模型证明的(275)。其他化合物可能会修饰KEAP1,使其不再能够在转录因子内的高亲和力和低亲和力位点与NRF2相互作用。值得注意的是,一些亲电的NRF2激活剂,如环氰烯酮,以共价形式与硫醇结合,但不形成不可逆的加合物(276,277)。因此,它们结合了不可逆共价药物(即高效力和持续的靶点参与)和可逆非共价药物(即缺乏永久性修饰,因此可能破坏或免疫原性其蛋白靶点)的理想特性(278)。这类分子的一个潜在缺点是,它们可能与KEAP1以外的蛋白质中对氧化还原敏感的半胱氨酸发生反应,从而影响其功能。巯基反应性分子的特异性问题表明,对于每种化合物,需要确定在其他蛋白质中不存在硫醇修饰的情况下允许治疗相关KEAP1修饰的窗口(图1b)。KEAP1内的靶半胱氨酸以及其他含巯基的非靶蛋白的这种分层呈现被称为“半胱氨酸代码”(279),并且受诱导剂的化学和剂量影响(280)。
KEAP1-NRF2 PPI抑制剂的一个重要潜在优势是提高了靶点的选择性。然而,对于任何小分子,不能完全排除脱靶效应。此外,KEAP1靶向NRF2以外的蛋白进行泛素化,这些蛋白也可能受到影响(281)。PPI抑制剂的脱靶选择性已被评估的几种类型之一是强效单酸性磺胺类;例如,化合物14似乎对KEAP1具有相当的选择性,在葛兰素史克公司(GlaxoSmithKline)增强的体外实验交叉屏幕面板中显示出适度的活性,以识别潜在的脱靶效应(246)。
动物模型。阻碍大多数慢性病药物开发成功的一个问题是动物模型中人类病理的不完全可复制性(288)。这一问题可能与人类退行性疾病缺乏一个非常重要的标志有关,即与NRF2相关的体内平衡功能的逐渐丧失。事实上,尽管在人类仍有许多工作需要做,但动物研究证据表明,NRF2活性随着年龄的增长而下降,并且NRF2的药理学或遗传学上调可延长寿命并改善健康状况(283, 284)。慢性疾病动物模型缺乏“NRF2变量”,这在AD小鼠模型中得到了很好的证明。目前的模型是基于这样的假设,即有毒的APP或tau突变蛋白在原本健康的动物背景下的表达复制了人类的病理。尽管这些模型提供了关于蛋白病的发病和进展的有价值信息,但是它们未能将治疗成功应用于人类。这种失败的原因可能与这些模型不考虑与NRF2相关稳态功能的疾病相关性下降这一事实有关。更复杂的临床前模型将需要一种反向翻译方法,这种方法不仅要在动物身上复制特定疾病的解剖病理特征,而且要复制受损的氧化还原、炎症或蛋白稳态。因此,NRF2敲除小鼠与AD患者甚至老年人有许多共同改变(156)。最近的研究表明,与野生型动物相比,喂食高脂肪食物的NRF2敲除小鼠表现出更大的神经血管功能障碍、血脑屏障破坏、神经炎症、淀粉样基因表达和认知能力下降,这与随着年龄增长而发生的许多表型变化类似(285)。引入低NRF2表达作为降低稳态反应的变量,可能有助于改进现有的慢性病模型,以获得更好的治疗效果。考虑到随着衰老NRF2活性的缓慢下降,杂合的NFE2L2 +/-小鼠可能是复制基础表达减少和药理学上调的合适模型。
癌症风险。从药理学角度来看,有必要确定NRF2水平升高超过致癌安全阈值的风险有多大(图4)。促进NRF2在癌细胞中稳定性的体细胞突变影响与NRF2的药理学激活引起的影响存在显著差异。因此,KEAP1和NFE2L2的体细胞突变导致NRF2活性升高而不受限制,这与NRF2激活剂的药理作用引起的脉冲激活完全不同。动力学特征,如曲线下的振幅和面积,反映NRF2信号的强度和持续时间,在基因激活设置和药理学手段方面有很大差异(202, 286)。此外,KEAP1基因丢失或失活突变似乎与该蛋白的药理学抑制作用不一样。因此,KEAP1丢失导致的NRF2活性以及E3连接酶适配器的几个致癌靶标(包括IKKβ和B细胞淋巴瘤2,bcl-2)增加,可能至少是恶性转化的部分原因。因此,在具有功能性KEAP1的一组细胞系中,成熟的NRF2激活剂,即BARD类似物RTA-405,增加NRF2但不增加IKKβ或BCL-2水平,并且不赋予肿瘤细胞生长或存活优势(287)。
虽然目前NRF2激活剂的癌症风险不能被忽视,但令人鼓舞的是,DMF III期试验荟萃分析显示安慰剂和DMF治疗组之间的癌症发生率没有差异(288)。 同样重要的是要注意,一些NRF2激活药物如果具有抗肿瘤作用的其他靶标可以降低这种风险。因此,最近发现DMF抑制GAPDH(289),并且这种抑制可能导致高度糖酵解的KRAS或BRAF突变癌细胞的能量危象,从而阻止肿瘤发展,如维生素C(290)。
展望
许多慢性疾病的标志是丧失稳态,如氧化还原信号、代谢灵活性、炎症控制和蛋白质稳态。这些复杂疾病的多因素性质可以通过对转录因子NRF2的单次靶向激活,产生有益的、全面的、多靶点和持久的细胞保护作用。尽管KEAP1-NRF2系统的特殊性使得这种方法在监测靶向参与和脱靶效应方面特别具有挑战性,但生物制药公司正在缓慢而稳定地开发针对NRF2主要调控因子KEAP1的药物。此外,需要明确排除增加癌症风险的担忧。在未来几年,正在进行的临床试验无疑将在回答这些问题方面取得重要进展。
图1:KEAP1对NRF2的调控及其药理靶标。
图2:KEAP1和NFE2L2 mRNA在人脑和血液中的表达。
图3:KEAP1结构域的晶体结构。
图4:根据NRF2的状态调节癌症风险。
本文由福山生物翻译整理,转载请注明出处。