






最新资讯



-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
含硒小分子作为潜在抗肿瘤药物的合成研究进展
发表于:2019-11-05 作者:admin 来源:本站 点击量:15438
摘要:含硒小分子由于他们各种不同的生物活性,已引起化学和医学研究人员的广泛关注。比如:抗肿瘤作用,心血管保护,抗菌或抗病毒作用,免疫调节和神经保护,其中最有前途的领域是抗肿瘤作用。在过去的几十年里,不同种类的有机硒化合物,如硒化物、硒酸盐(iso)、取代硒脲、硒酯和含硒杂环化合物被报道作为抗癌药物的候选。目前对含硒抗癌化合物的研究主要集中在对其生物活性的研究,而对其合成方法的研究较少。本文综述了近年来合成具有较强抗肿瘤活性的有机硒化合物的方法,为进一步设计和合成具有不同结构特征的新型含硒生物活性分子提供参考。
关键词:抗肿瘤,硒,合成方法,抗肿瘤药物,癌症,分子。
1.简介
在过去的几十年里,癌症一直被认为是一个主要的公共健康问题。目前,它已成为仅次于心血管疾病的全球第二大死因。根据《2014年癌症报告》,2012年全球约有1410万新病例和820万与癌症相关的死亡。与2008年的1200万和700万相比,这一数字有了大幅增长。此外,治疗癌症的成本是巨大的。2009年,新增癌症个案的医疗开支估计为2,860亿元。这个数字如果将纳入治疗后的癌症预防和康复成本考虑在内,则会变得高得多。在过去的几十年里, 化疗药物的开发已经取得了相当大的进展。许多药效更强副作用更小的药物已经开发出来。尽管制药业取得了一定的成就,但选择性和耐药性仍然存在,仍然是研究人员面临的最艰巨的任务。
虽然硒、氧和硫都属于氧族元素,但含硒化合物中键强度较弱,键长较长,具有独特的化学反应特性。例如:在依布硒林的合成中,二硒类化合物和氯化亚砜反应可以得到氯硅烷。这种反应在相应的二硫键中很少见。硒也是一种重要的微量元素,具有多种生物活性。含硒分子已被证明对肿瘤、心血管疾病、病毒或细菌感染、免疫相关疾病和神经降解疾病有效。
在这些适应症中,最有前途的药物治疗领域是抗肿瘤药物。关于硒抗癌活性的最早研究可以追溯到20世纪60年代,当时科学家发现硒的分布不均匀,生活在富硒地区的人患癌症的几率往往较低。这些独特的反应特性以及令人惊讶的生物活性激发了研究人员设计不同类别的含硒化合物用于癌症治疗。近几十年来,合成了多组有机硒化合物,并对其抗肿瘤活性进行了深入研究。
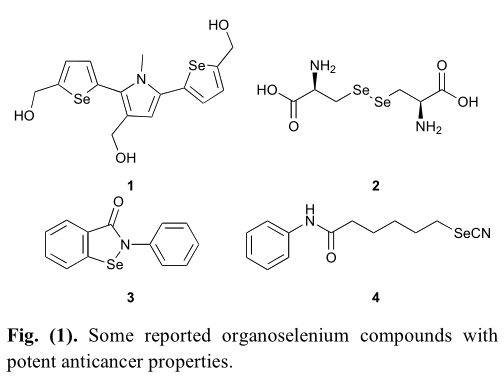
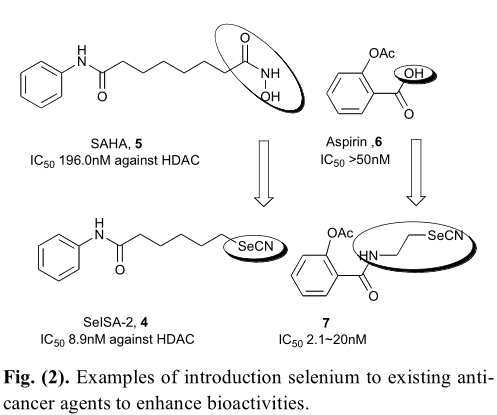
在所有合成特征和硒有机化合物,其中一些已被证明是有效的对几种类型的癌症细胞系,如结肠癌细胞系(HT29),乳腺腺癌细胞系(mda-mb-231),胰脏癌细胞系(PANC-1),恶性黑色素瘤细胞系(UACC903)(图1)[3]。然而,这些化合物的抗癌作用机制通常仍不清楚。根据文献,硒可能通过两种不同的途径抑制癌细胞的生长。在正常细胞中,硒可以作为一种抗氧化剂帮助这些细胞抵御氧化损伤,而在肿瘤前细胞中,硒会转化为一种预氧化剂,使这些细胞更容易受到硒的攻击。因此,据报道,肿瘤细胞一般更容易受到硒化合物所表现出的细胞毒性作用的影响。此外,有机硒化合物的抗癌作用还可通过细胞凋亡、抑制血管生成、调节血管活性等途径实现。基于这些发现,考虑将硒与化疗或化学预防药物结合,比如SAHA和阿司匹林等,提供新的含硒化合物具有更强的或更好的抗肿瘤药效果被认为是一种很有前途的方法选择性识别新的抗癌药物类别(图2)。
迄今为止,已报道了几种结构特征各异的有机硒化合物。目前对这些化合物的研究主要集中在其生物活性方面[20-22]。从有机合成的角度,我们认为,系统地总结合成具有强大抗癌性能的有机硒化合物的方法,对于进一步研究发现新的含硒肿瘤治疗衍生物也具有重要意义。本文从含硒有机分子的结构特点出发,对近年来报道的含硒有机分子进行了分类,并对其合成方法进行了综述,对今后的新型含硒生物活性小分子的设计、合成及结构修饰的研究具有一定的指导意义。
2.不同类型有机硒化合物的合成方法
2.1 硒氰酸酯和硒异氰酸酯
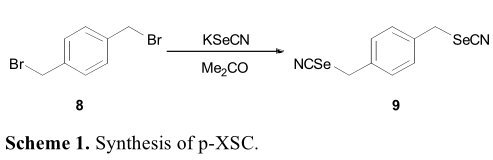
硒氰酸酯和硒异氰酸酯在肿瘤中对多种信号通路具有抑制作用。甘瑟首次报道了将硒氰酸盐引入有机化合物,硒氰酸钾(KSeCN)作为亲核试剂与1,4-二溴甲基苯(8)的偶联反应得到1,4-双(硒氰基-甲基)苯(9)(p-XSC),它是一种强化学预防药理含硒化合物(方案1)。此后不久,Ghose对人口腔癌进行p-XSC的检测,发现其化学预防作用是由丝裂原激活蛋白激酶和Fas通路诱导的。
硒氰酸酯与现有抗癌药物最著名和最成功的组合之一可能是SAHA的结构修饰,SAHA是一种现有的组蛋白去乙酰化酶抑制剂。硒氰酸盐与SAHA的结合显著增加了对组蛋白去乙酰酶的抑制活性(方案2)。含硒阿司匹林衍生物7,其IC 50值达到2.1-3.4 nM,是氟脲嘧啶(5-FU)治疗结肠癌阳性对照(方案2)[19]的10倍。此外,它对正常细胞的细胞毒性也降低了,表明增强了安全性。Roy和他的同事发现2-(5-溴戊基)-7-氨基苯并(de)索奎诺林-1,3-二酮(14)对转化细胞具有潜在的抗血管生成和细胞毒性作用。此外,他们也试图将硒氰酸酯部分移植到该结构上,以进一步提高生物活性(方案2)。对合成的硒氰酸酯衍生物(16)的机制研究表明,其抗癌作用可能是通过p53依赖性凋亡诱导的。对于硒异氰酸酯的构建,传统的策略是霍夫曼碳胺反应。KOH和三氯甲烷处理初级胺时,原位生成卡宾结构,在回流的THF中,硒和三乙胺反应生成相应的硒异氰酸酯。也有报道称,光气可用于生成异氰酸酯,并可进一步转化为硒异氰酸酯(方案3)。
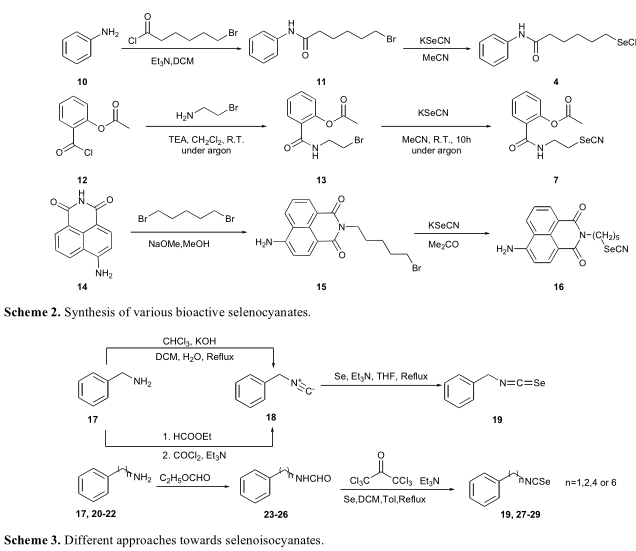
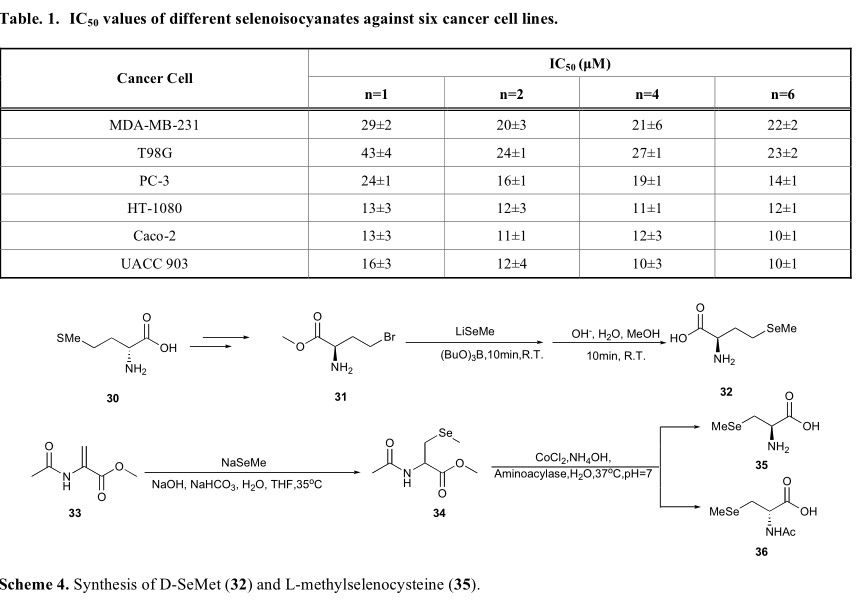
Fernandez-Bolanos报道了一种一锅法合成硒异氰酸酯的方法。苯烷胺类甲酰化后,相应的甲酰胺经过三光气和硒的反应,得到相应的硒异氰酸酯的衍生物19和27 - 29(方案3)。该系列化合物的生物活性评价表明, 硒异氰酸酯可以抑制黑色素瘤细胞生长,前列腺癌和乳腺癌等IC 50值达到10-40μM,如表1所示,稍微高于相应的异硫氰酸酯,这些异硫氰酸酯的IC50值从20~50μM 不等。这种一锅法的策略是很好的,由于气相光气可以被三光气所取代,因此传统的气相光气合成方法难以实现。此外,硒异氰酸酯可以以甲酰胺为原料一步合成。
2.2 硒化物
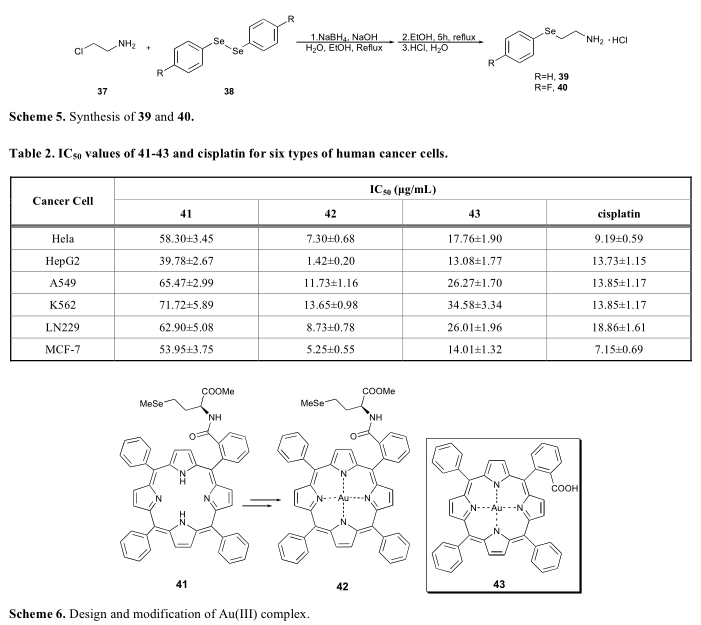
硒化物结构存在于许多含硒氨基酸衍生物中,如硒代蛋氨酸(Se-Met, 32)、硒二谷胱甘肽(SeGSH)和甲基硒半胱氨酸(35),目前被广泛认为是硒在哺乳动物中可能的存在形式(方案4)。然而,据我们所知,SeGSH的合成尚未见报道。这些含硒氨基酸在体内外均经过检测,对不同的癌细胞系如人乳腺癌细胞、人结直肠癌细胞等均表现出较强的抑制作用。由于该类分子通常具有手性中心,因此构建含硒氨基酸的合成方法有两种。一种方法以纯对映体为原料。例如,Cao从d -蛋氨酸(30)经几个步骤合成了31。甲烷硒酸锂被用来将单硒官能团引入31(方案4)。
另一种方法是将含硒氨基酸作为外消旋体混合物,通过水解酶进行拆分,具有较高的手性选择性。例如,Liu报道了一种合成L-甲基硒半胱氨酸的方法(35)。首先通过33与甲硒酸钠(NaSeMe)反应合成了一对硒半胱氨酸对映体。采用L-氨基酰基酶催化水解,分离出两种对映体(方案4)[41]。
Kang报道了苯氨基乙基硒化物39和40在体内和体外对前列腺癌均有抑制作用(方案5)[42]。该反应的机理可能与二硒酰胺的还原有关。原位生成的还原硒可以作为亲核试剂进攻37生成目标化合物39和40。
铂(II)顺铂、卡铂、奥沙利铂等配合物药物在癌症治疗中应用广泛。由于与Pt(II)配合物的结构相似,Au(III)配合物在这方面也受到了广泛的关注。有报道称,与顺铂相比,部分Au(III)配合物对不同的癌细胞株具有更强的抑制作用,说明Au(III)配合物也可以作为一种新型的抗癌药物系列[43]。Ruan在前人研究的Au(III)衍生物43的基础上,设计合成了一种硒修饰的卟啉Au(III)复合物42[44]。机理研究表明,42通过诱导线粒体依赖的凋亡和抑制细胞周期而表现出较强的细胞毒性(方案6),硒修饰的卟啉Au(III)复合物的细胞毒性作用强于原Au(III)复合物(43)和顺铂,如表2所示。
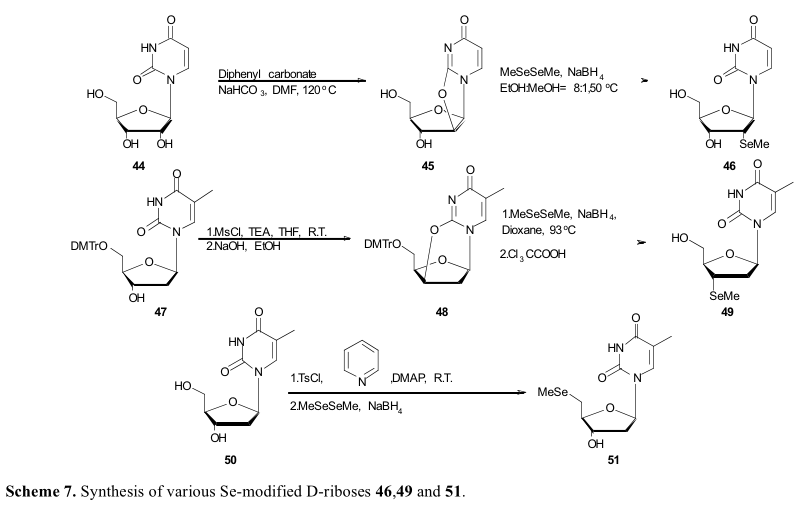
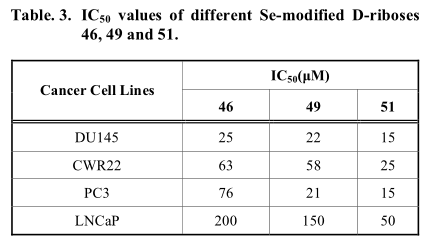
甲基硒取代基也被引入核苷结构。林报道2 ',3 ' - 5 ' –Se修饰D一核糖衍生物(46,49和51)能够诱导半胱天冬酶的细胞凋亡在不同肿瘤细胞系(表3)。在他们的合成策略中, 化合物44的2,2’分子内环合是在碳酸二苯酯和碳酸氢钠存在的条件下,在DMF中反应得到2,2’-无水尿苷 (45),然后再经过二甲基联硒化物和硼氢化钠的处理得到46。该策略对3 ' -甲基胸苷(49)的合成也很有效。然而,在2,3 ' -分子内环化之前,需要通过甲基化反应激活3 ' -羟基。48的后续开环反应在相似的反应条件下进行,但温度较高。林将这种现象归因于环应变较小,与45相比。对甲苯磺酰酯别选择性的引入到化合物50中5′c位置作为活性离去基,构建5′-甲基硒尿苷。然后,在硼氢化钠存在的条件下,所得到的甲苯磺酸酯与二甲基二硒发生偶联反应,形成单硒酰胺(Scheme 7)。
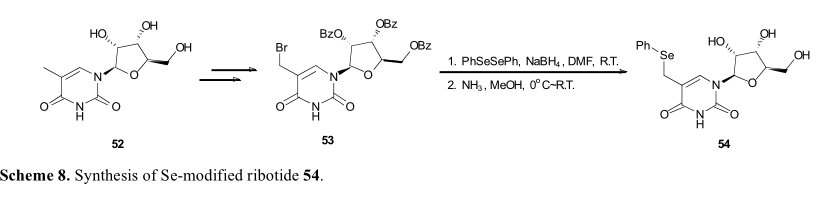
Greenberg及其同事报道,在碱基片段引入硒也可以实现核苷的硒修饰。在这种情况下,糖基上的羟基应首先得到充分保护。52经过NBS的介导提供烯丙基溴,然后用二苯基二硒酰胺和硼氢化钠处理,得到单硒酰胺(54),过程与上面讨论的类似(图8)。
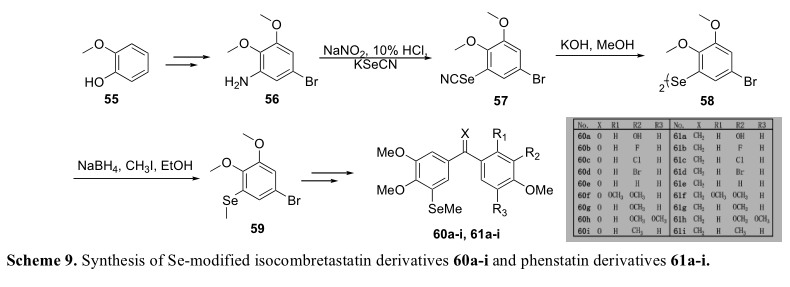
芳硒的合成方法通常涉及重氮盐的形成。例如,最近设计合成了一系列甲基硒与异共促食素和苯他汀衍生物的连接物。在这项工作中,作者打算用甲基硒取代天然产物isoCA-4中的C-3、C-4和C-4’的甲氧基,发现C-4的取代几乎不可能发生。根据他们的解释,两个邻位甲氧基可能抑制反应活性。因此,他们试图将甲基硒连接到C-4’位置,尽管结果显示衍生物的细胞毒性并不比异辛卡-4高。基于这些发现,他们主要关注C-3取代。本系列中活性最强的分子61a对不同的癌细胞株具有抑制作用,其IC50值为2-11 nM。(表4)力学研究表明,61a具有管蛋白的功能,就像isoCA-4一样。在合成过程中,重氮盐作为离去基引入硒氰酸盐基团。合成的硒氰酸酯(57)经硼氢化钠还原,随后与碘甲烷进行甲基化,得到甲基(芳基)硒片段。(方案9)Wen报道了一系列含硒/硫的a-4类似物,并检测了它们对SGC7901、HT1080和KB细胞株的生物活性,见表5[48]。用硒取代吲哚结构取代了CA-4的B环和顺式双键。以1,2-双(3,4,5-三甲氧基苯基)二硅烷(64)为硒源,通过重氮盐63与二硅烷钠的偶联反应得到。在三氯化铁和碘的存在下,二硒酰胺键断裂,在微波反应下把吲哚基部分连接起来。(表10)。

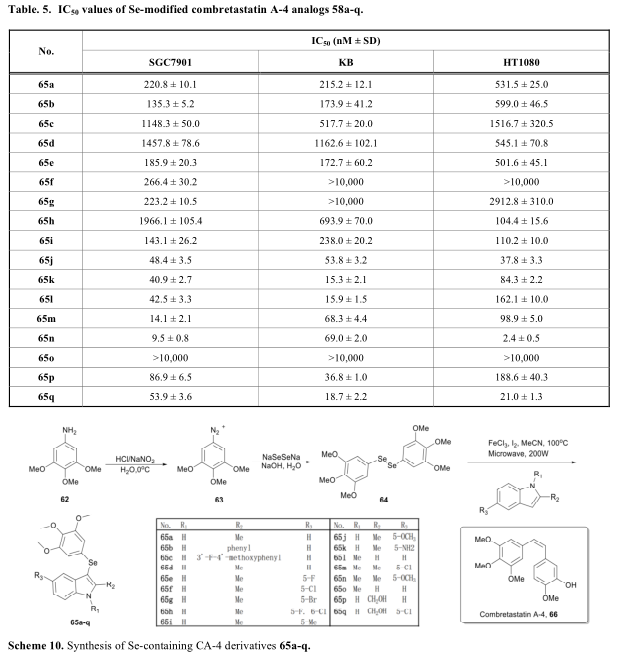
Moreno报道了一系列含硒嘧啶衍生物[2,3-d],在体外显示了强大的细胞毒活性(表6,方案11)[49]。与一般构建硒化芳基的方法不同,他们通过硒异氰酸酯和邻氨基芳腈的环化或用硒脲亲核取代氯基,成功构建了含硒嘧啶[2,3-d]。硒元素的引入不需要重氮盐的形成。在生物活性研究中,他们还测试了其中间体硒醇68,73和74的细胞毒活性并发现68a (X=C)也具有较强的细胞毒活性。
2.3 联硒化物
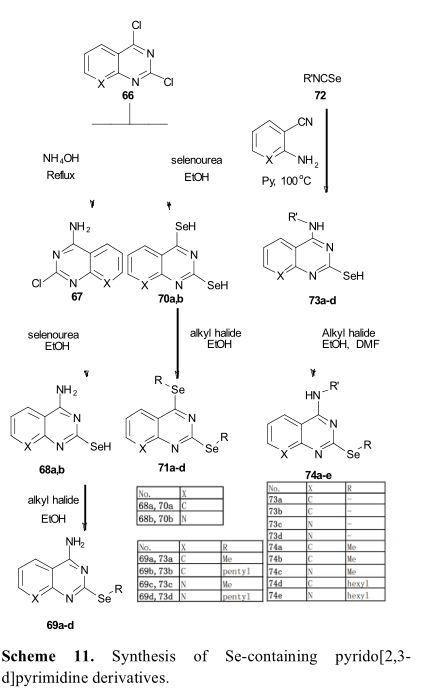
取代二硒醚类化合物由于在小鼠肝脏、肾脏和大脑中易于分布,已在生物系统中显示出有趣的药代动力学特性。因此,取代二硒醚类化合物有潜力开发成为有前途的抗肿瘤药物脑癌或肝癌。Battin公布环硒二聚体(2),拥有强大的对不同细胞DNA损伤抑制IC 50值变化从3.34微摩尔到25.1微摩尔 (Scheme12) [51]。
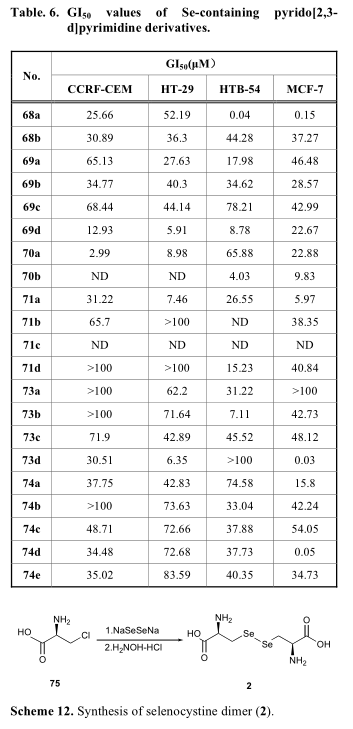
对于有机硒分子中二硒酰胺结构的合成,通常采用Na2Se2或Se3CN2作为硒源[51-56]。与单硒类化合物的生成不同,单硒类化合物的生成通常涉及到小二硒类化合物的原位还原,二硒类化合物的合成可以通过直接亲核攻击实现,从二硒类化合物(如Na2Se2或Se3CN2)到另一个带有活性离去基的偶联(方案13-15)。
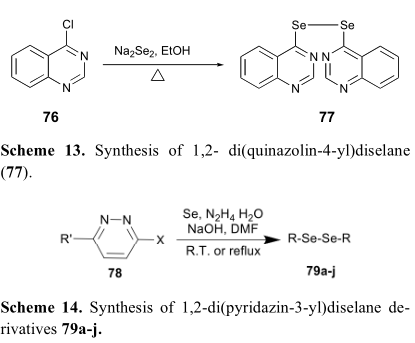
采用类似的策略,通过Na2Se2或[Se+ N2H4]在芳香环上取代活性氯来构建芳香二硒醚类化合物(方案13-14)。黄合成的1,2-二(喹唑啉-4-酰基)二硒醚(77)对不同的癌细胞如A549、PC-3、MDA-MB-435等具有广谱的抗癌作用。
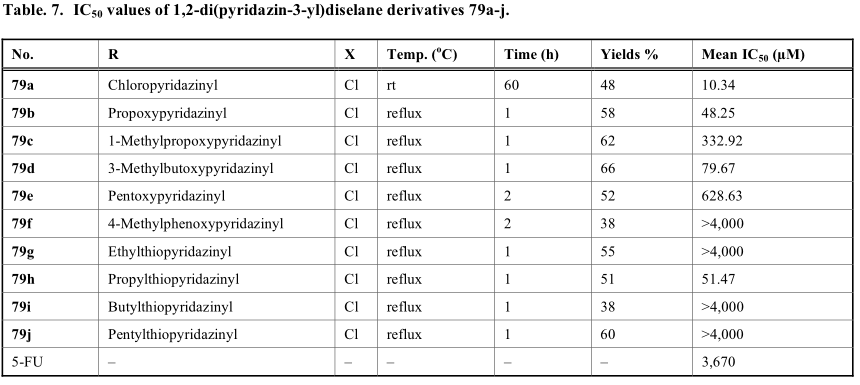
由金合成的一系列1,2-二(吡啶-3-酰基)二硒醚衍生物(79a-j)具有对MCF-7的抗多聚性活性,如表7所示。根据文献,有几种不同的合成策略来构建对称二硒醚类化合物(方案15)。例如,用苯基氨基与作为硒源的Se3CN2反应得到中间产物硒氰酸酯衍生物(80)。随后,对关键中间体80进行硼氢化钠的还原,得到了理想的二硒酰胺化合物[54,56]。谷口教授开发的另一种策略依赖于芳基碘化物(82)与硒在铝、2,2'-双吡啶和碘化亚铜[55]存在下的直接耦合反应。虽然这种方法一般进展缓慢,但可以避免使用硼氢化钠等还原剂。
2.4 亚硒酯类
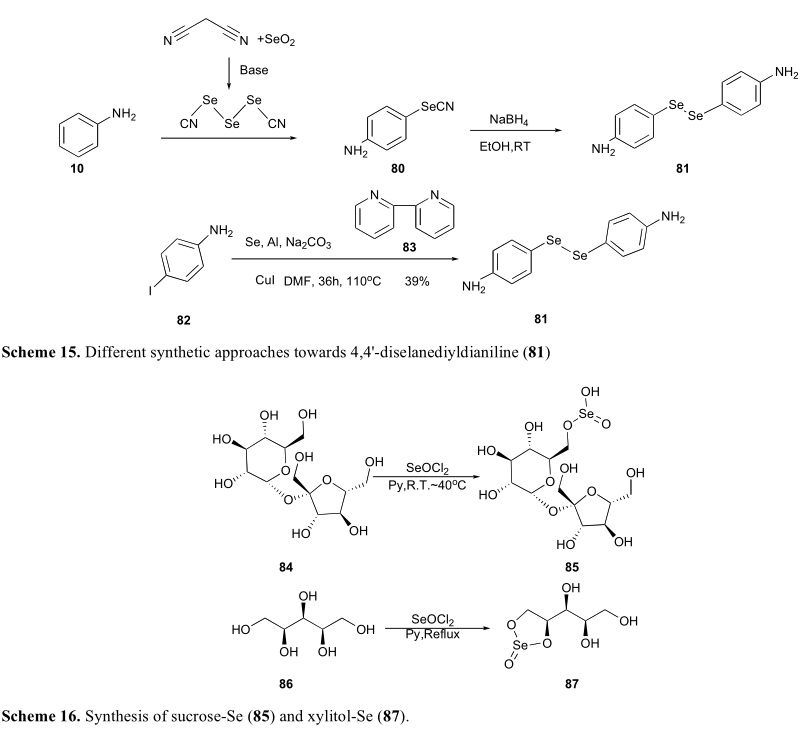
最近,郭和他的同事将硒作为硒酯引入碳水化合物中。用二氯化亚硒酸盐处理不同糖时,观察了区域选择性酯化反应。对于蔗糖,可以得到单酯(85)。当二氯化亚硒酸盐应用于木糖醇时,得到了五元环亚硒酸酯(87)(方案16)。85和87对不同的癌细胞株均有较好的抑制作用。
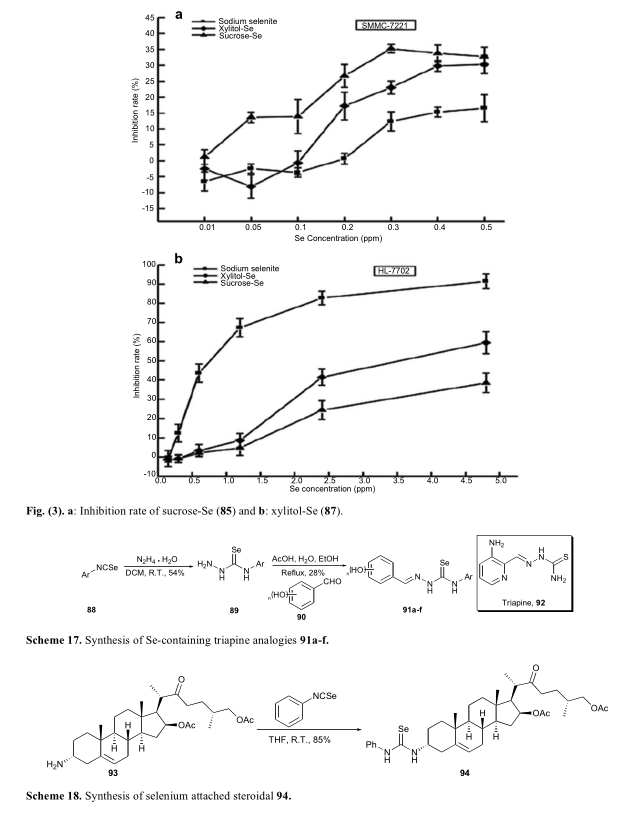
其中最常用的无机硒源之一亚硒酸钠已被用作营养补充剂。近年来,亚硒酸钠也被用于治疗癌症。但有报道指出,亚硒酸钠的毒性作用不容忽视。亚硒酸钠的进一步研究表明,Na2SeO 3在极低浓度下对癌细胞(SMMC-7221)和正常细胞(HL-7702)均表现出明显的细胞毒性, 85和87可选择性抑制SMMC-7221在0.15 ~ 1.2 ppm Se的检测浓度下,表现出较高的选择性[57,58]。
2.5 取代的硒脲
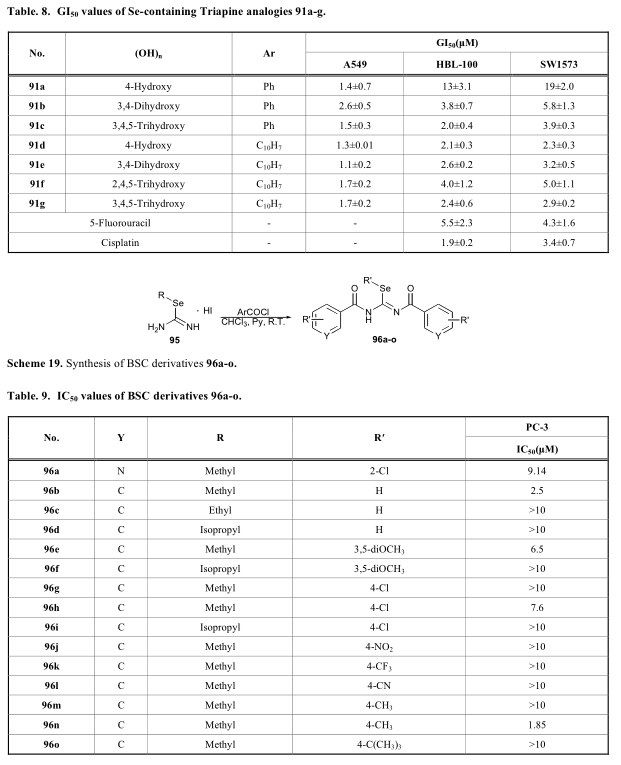
取代硒通常被引入抗氧化分子中,以进一步增强其生物活性[60-62]。据报道,含硒脲结构的化合物可作为活性氧(ROS)的清除剂,是一种很有前途的抗癌策略。Calcatierra受席夫碱衍生物triapine(92)的启发,报道了一系列硒脲衍生物(91)具有抗氧化、清除活性氧等多种生物活性,如表8[60]所示。Laura将硒脲与一些内源性的生物活性甾体结构连接起来,在进一步的生物活性测试中,结果显示含硒衍生物(94)清除活性氧增加,对癌细胞具有协同作用[61](方案17,18)。
Plano报道了一系列对PC-3细胞系具有较强细胞毒性作用的双嘧磺酰氨基甲酸盐(BSC)衍生物,见表9[63]。本系列中对PC-3最有效的衍生物96n的作用机制研究表明,BSCs治疗肿瘤细胞的凋亡可能是由致死的有丝分裂诱导的。与上述硒脲类衍生物不同,BSCs的合成往往是从各种烷基氨基咪唑烷酸氢碘化物开始的(95)和不同的酰氯,在吡啶作为催化剂的存在下,在室温下以1:2.1摩尔的比例反应(方案19)。
文献表明,二苯基取代硫脲是一种强的碳酸酐酶抑制剂,可在低氧条件下阻断肿瘤细胞的生长[64]。最近,Fabrizio Carta及其同事报道了一系列取代硒脲类化合物在常氧和低氧条件下对hCA亚型的抑制作用以及对PC-3、MDA-MB-231和HT-29的活性(Scheme 20)[65]。在PC-3和MDA-MB-231, 98a和98b中,在常氧条件下表现出与对照化合物相同或更强的抑制作用。三种化合物对HT-29均无活性(图4)。
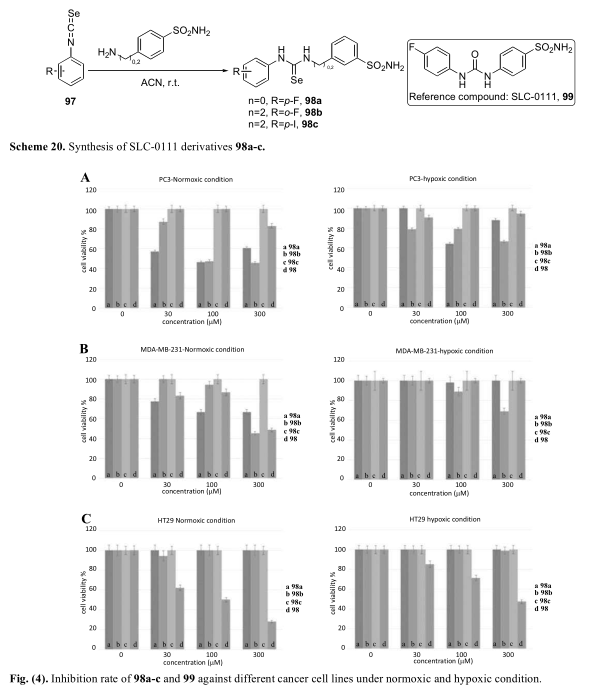
然而,所得的衍生物对不同类型的CA异构体没有很好的选择性。由于CA I和CA II广泛分布于健康细胞中,这些衍生物可能对正常细胞产生影响。因此,可能需要对这些含硒化合物进行进一步的安全性研究。
2.6硒代亚砜
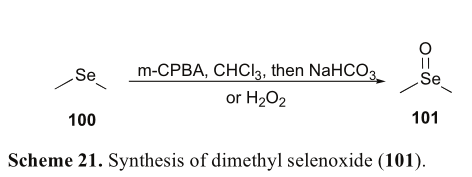
硒氧化二甲酯(101)是一种最简单的含硒生物活性分子,对人前列腺癌细胞株(LNCaP)具有较强的细胞毒作用[66]。随后,对其他癌细胞株的抗癌活性进行了测试。例如,二甲基氧化硒进一步检测对乳腺癌细胞(mda - mb - 231)与IC 50值为0.105μM [67]。治疗后的二甲基硒氧化物,即使在低剂量,两种癌症细胞活力下降[68]。硒化二甲酯的形成是通过m-CPBA或过氧化氢介导的二甲基硒过氧化反应完成的(方案21)[69]。
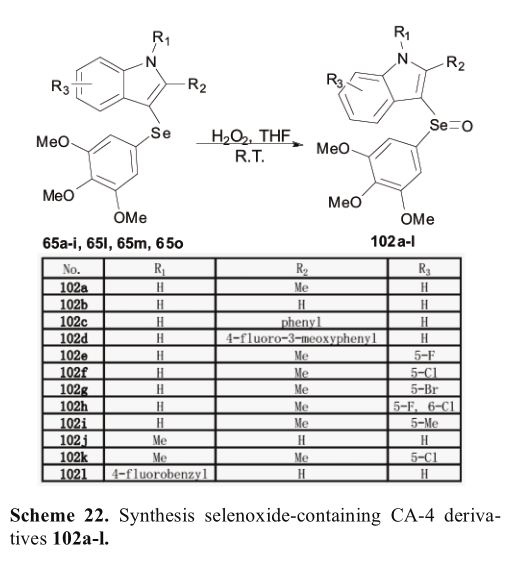
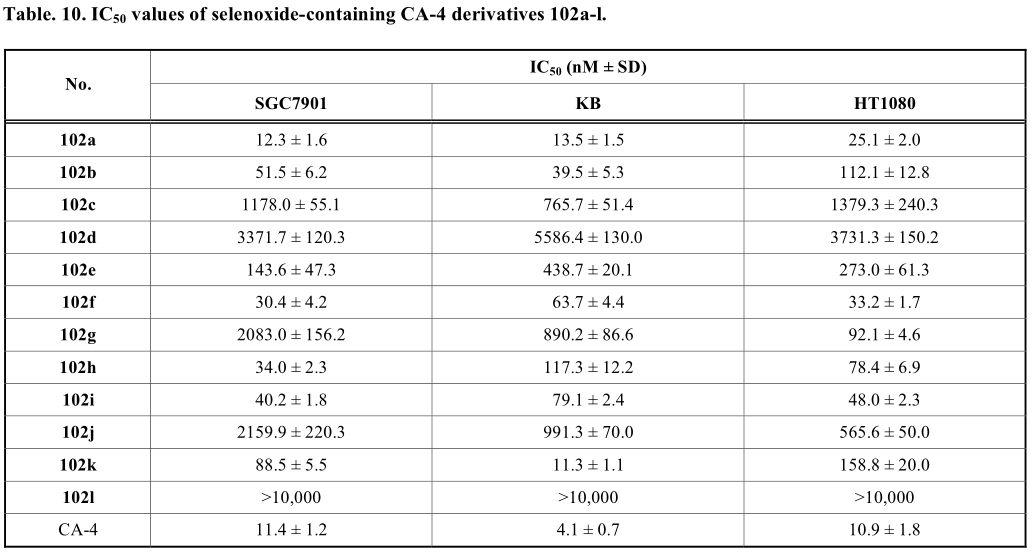
通过对单硒烯类化合物的制备和生物评价,文进一步合成了其硒氧化物类似物[48]。根据他们的方法,过氧化氢被进一步用于硒化物衍生物的氧化,得到硒氧化物衍生物(方案22)。一般来说,含硒氧化物连接剂的化合物表现出较弱的抑制作用,除了102a。(表10)
2.7含硒的杂环化合物
由于杂环结构的多样性,硒元素的引入可以提供不同的含硒杂环,具有不同的生物活性。含硒杂环化合物的合成策略因杂环结构的不同而异。
2.7.1 硒恶唑烷酮衍生物
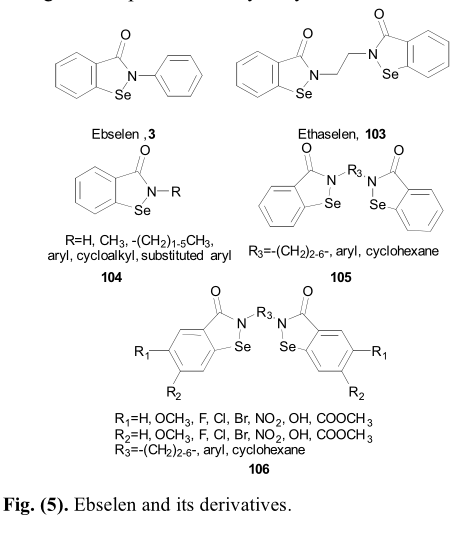
临床试验中第一个硒唑烷酮衍生物是依布硒林 (3),它首先被用作抗氧化剂,并被用于治疗噪音引起的听力损失。对晚期癌症化疗致听力损失的防治也进行了的研究。另一个衍生物,1、2 - (双 (1, 2-苯硒唑烷-3 (2 氢)酮))乙烷,名为乙烷硒啉(103)随后被设计合成,作为一种治疗胃癌、肺癌和结肠癌的有效的硫氧还蛋白还原酶抑制剂。Li报道了一系列硒唑烷酮衍生物(103-106)作为强效硫氧还蛋白还原酶抑制剂,有望进一步开发成为新型抗癌药物(图5)[70]。
构建硒唑烷酮结构的关键中间体为2-(氯代羰基)苯基次氯酸盐(109),用氯化亚砜对2,2'-异丙二基二苯甲酸进行处理可得到该结构。2-(氯代羰基)苯基次氯酸盐(109)与不同胺的偶联反应提供了多种具有硒唑烷酮结构的依布硒啉衍生物,其产率极好(图23)[12,71]。
最近,Sucheck和他们的同事开发了一种针对依布硒啉及其衍生物的新方法[72]。他们描述了硒氰酸钾(KSeCN)与N-取代的邻卤代烷之间的一种新的光热诱导铜介导的交叉偶联反应,形成2-烷基-1,2-苯并异硒噻唑-3(2H)-酮(Scheme 24)。有趣的是,在加热过程中,铜配体(1,10-菲罗啉)通过原子转移(AT)机制促进了C - Se键的形成。另一方面,在没有配体的情况下,光诱导活化可能通过单一的电子转移(SET)机制进行。根据他们提出的反应机理,AT涉及卤化物原子从芳基卤化物转移到a (phen)CuI (SeCN)络合物,形成笼状芳基(Ar·)和(phen)Cu II (SeCN)X络合物。进一步的偶联反应得到了芳基硒氰酸酯和(phen) cuIx,它可以通过配体交换再生(phen) cuI (SeCN)来完成这个循环。
芳基硒氰酸酯可以环化形成依布硒啉衍生物。一套光诱导的机制需要激进的亲核芳香取代反应(S RN1)和涉及光激发(Cu(SeCN)2)离子—提供一种激发态的(Cu(SeCN)2)−•自由基,可以形成一对激发态的[Ar-X•和CuII (SeCN)2]。随后自由基对的偶联过程提供了芳基硒氰酸酯和[Cu I (SeCN)X]−,它们可以进一步进行配体交换来完成催化循环。
2.7.2. 硒唑衍生物
Srivastava首次报道了一种1,3-硒唑衍生物(117),在体外对P388和L1210细胞具有新的细胞毒性,对小鼠Lewis肺癌具有抗肿瘤活性[73]。硒唑环是由氰化物(113)和3-溴-2-草酸乙酯(114)在Al2 Se3作为硒源的存在下耦合反应而成(方案25)。
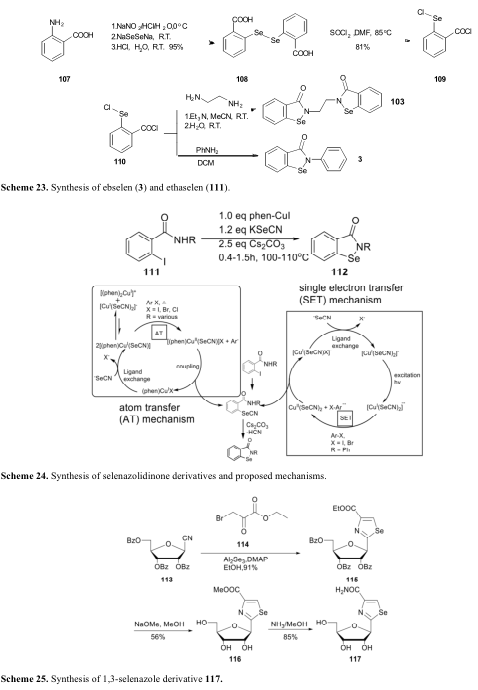
2015年,Zhou报道了一系列含有1,3-硒唑骨架的生物活性分子(119),经体内实验证明,这些分子对人乳腺癌MCF-7细胞有效(表11)[74]。进一步的机制研究表明,119g可通过减少ROS的生成诱导MCF-7细胞凋亡。硒异辛酸盐(118)是根据2.1节讨论的合成方法首次制备的,与2-氯吡啶-3-胺偶联得到硒唑衍生物119a-g(方案26)。
2.7.3. 硒酚衍生物
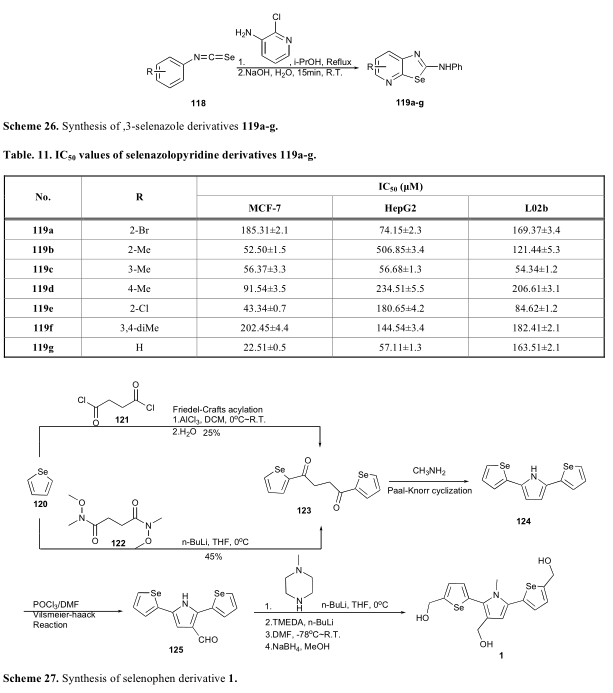
Juang报道一种包含硒酚的新型α三重杂环化合物(1),表现出对几种人类癌症细胞系有抑制活性与IC 50值的范围[75]。该化合物是以硒酚为原料合成的。硒酚首先用琥珀酰二氯(121)或N,N-二甲氧基-N,N-二甲基琥珀酰胺(122)处理,得到1,4-二酮衍生物(123)。随后1,4-二酮衍生物的PaalKnorr环化反应生成吡咯衍生物(124),然后进行Vilsmeier-Haack反应,在吡咯环上引入醛基。用n-BuLi和DMF对124进行处理,在硒吩基上又附加了两个醛结构。最后,硼氢化钠介导的还原反应将三个醛基转化为甲醇基(方案27)。
2014年,Arsenyan报道一系列硒酚 [3、2 c]和[2,3 c]喹诺酮衍生物, 对几种细胞系具有强烈的细胞毒性和IC 50值从6到100μg /毫升不等。不久之后,他们合成了硒苯[3,2-c]和[2,3-c]香豆素衍生物,具有一定的细胞毒性[77]。最近,他们利用这一策略合成了更多的类似物(128a-f, 132a-f),并进行了进一步的生物学研究。[78]机理研究表明,该系列化合物能够抑制特定的抗氧化酶系统,如SOD、GPx(选择性抑制肿瘤细胞GPx活性)和TrxR;降低ROS浓度。在他们的合成过程中,建立了一个共轭烯烃-炔形成了硒酚骨架。随后,原位生成SeBr4(由二氧化硒和浓氢溴酸制备)引发分子内环化,得到了目标硒酚衍生物。
2.7.4. 硒二唑衍生物
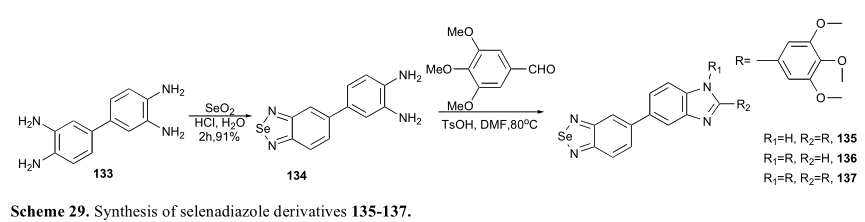
有报道称1,2,5-呋喃嘧啶是小分子表达抗癌活性的重要结构基序[31,79 -81]。1,2,5-硒代二唑可视为硒代氧氟杂氮。因此,设计并合成了多种含硒二唑骨架的分子。他们的生物活性测试得到了令人兴奋的结果。例如,Liang合成并评价了一系列硒二唑衍生物(135-137),它们可以诱导不同癌细胞株caspase和p53依赖的凋亡。肿瘤细胞在x射线照射下(8 Gy)细胞毒作用显著增强,说明该系列化合物可增强肿瘤细胞的放射敏感性(图6)[80]。
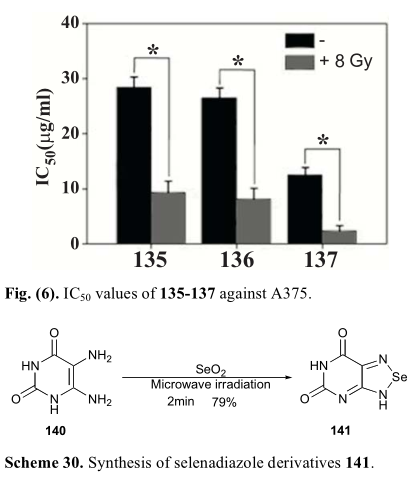
构建苯并苯二唑骨架的一般步骤是邻二胺与SeO2的偶联反应(方案29-31)。结果表明,微波辅助反应可以使这种转化更加有效。并且可以在较短的反应时间内得到目标硒代唑,收率较高(方案30)。张和他们的同事设计并合成了一系列含硒二氮唑的CA-4衍生物。构建硒二唑环可以在结构中引入硒元素,在一定的时间内,还可以构造出有机硒CA-4衍生物的构象。在本系列中,143a/b化合物中有两种具有比CA-4更强的细胞毒活性,如表13所示。在本例中,除邻二胺外的邻二酮肟(142)被用作偶联反应物之一(方案31)。
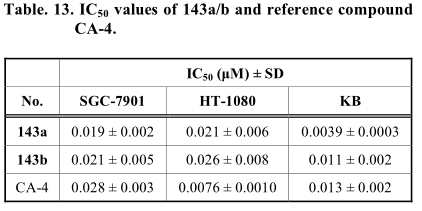
Arsenyan报道了一系列具有不同酰胺取代基的1,2,3-硒二唑衍生物[82]。进一步对这些化合物在HT-1080(人)和MG-22A(小鼠)上的体内生物学评价表明,这些化合物具有强大的抗肿瘤活性。1,2,3-硒二唑的形成与乙基乙酰乙酸氨基脲(144)与二氧化硒的环化反应有关。然后在室温将所得酯(145)转化为不同的酰胺,得到化合物146-149(方案32)。这些反应均在黑暗中进行,以防止硒二唑循环光降解。
2.7.5. 四氢化硒酚衍生物
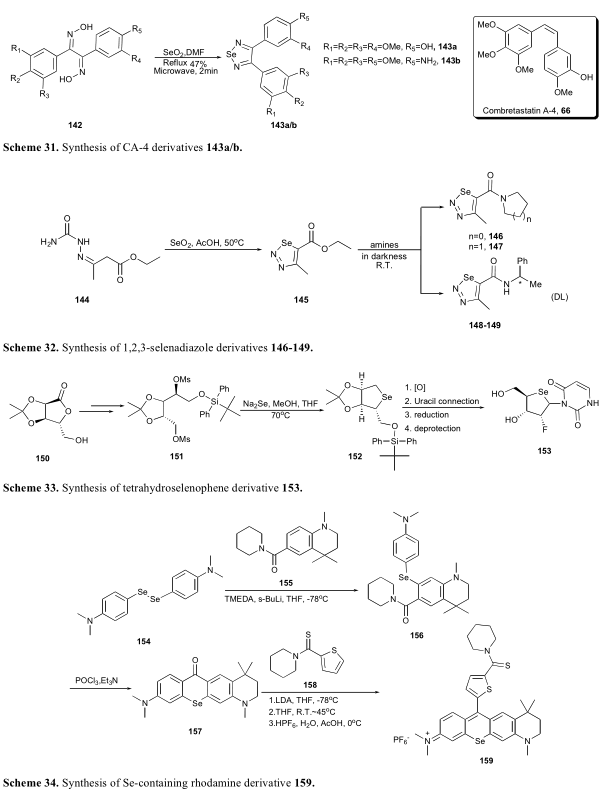
由于硒和氧的性质相似,核苷酸衍生物的一种实用的结构修饰策略是用硒原子取代糖骨架上的氧原子。按照这一策略,Kim报道了一系列含硒核苷酸[83]。其中一个最强大的衍生品153年可能抑制HCT116、A549, SNU638, T47D和PC-3细胞系在浓度为1微摩尔时。合成方法中关键的环化反应是Na2Se和甲磺酸盐的连续亲核取代。合成的中间产物152经过硒氧化、尿嘧啶连接减少和去保护得到153(方案33)。
2.7.6.硒罗丹明衍生物
Hill报道了一种罗丹明衍生物(159),可作为光敏剂用于表达p- gp的癌细胞的光动力学治疗[84]。这种硒罗丹明衍生物提供了一个固氮原子与黄嘌呤核结合的理想光谱值,可以有效地对抗p- gp表达细胞。154和155的偶联反应在s-BuLi和TMEDA存在下进行。在乙腈中,在POCl3 /Et3N存在下,硒化二芳基(156)进行了后续环合反应。最后,在HPF6中,化合物157和158的偶联反应得到了硒罗丹明衍生物(159)(Scheme 34)。
结论
迄今为止,含硒分子的设计通常遵循两种不同的策略。其中一种采用了生物同分异构的概念,即在生物活性化合物中以氧或硫原子取代硒原子。另一种方法是以硒化物、硒(异)氰酸盐的形式引入硒基,取代硒脲或硒酯来报道生物活性分子或批准的抗癌药物,目的是进一步增强其生物活性或提高细胞毒性选择性。
一般来说,由于不同含硒分子结构的差异,构建不同含硒分子的合成方法也不尽相同。硒源既可以作为亲电试剂,如元素硒,也可以作为二烯类化合物或亲核试剂,如硒氰酸盐或甲烷烯醇酸盐。对于含硒杂环,硒源对环化反应的参与程度因杂环结构骨架的不同而异。本文综述了近年来含硒抗癌药物的合成方法,希望对今后新型含硒抗癌药物的设计和合成有一定的指导意义。
本文由福山生物整理翻译,转载请明确注明出处。
关键词:抗肿瘤,硒,合成方法,抗肿瘤药物,癌症,分子。
1.简介
在过去的几十年里,癌症一直被认为是一个主要的公共健康问题。目前,它已成为仅次于心血管疾病的全球第二大死因。根据《2014年癌症报告》,2012年全球约有1410万新病例和820万与癌症相关的死亡。与2008年的1200万和700万相比,这一数字有了大幅增长。此外,治疗癌症的成本是巨大的。2009年,新增癌症个案的医疗开支估计为2,860亿元。这个数字如果将纳入治疗后的癌症预防和康复成本考虑在内,则会变得高得多。在过去的几十年里, 化疗药物的开发已经取得了相当大的进展。许多药效更强副作用更小的药物已经开发出来。尽管制药业取得了一定的成就,但选择性和耐药性仍然存在,仍然是研究人员面临的最艰巨的任务。
虽然硒、氧和硫都属于氧族元素,但含硒化合物中键强度较弱,键长较长,具有独特的化学反应特性。例如:在依布硒林的合成中,二硒类化合物和氯化亚砜反应可以得到氯硅烷。这种反应在相应的二硫键中很少见。硒也是一种重要的微量元素,具有多种生物活性。含硒分子已被证明对肿瘤、心血管疾病、病毒或细菌感染、免疫相关疾病和神经降解疾病有效。
在这些适应症中,最有前途的药物治疗领域是抗肿瘤药物。关于硒抗癌活性的最早研究可以追溯到20世纪60年代,当时科学家发现硒的分布不均匀,生活在富硒地区的人患癌症的几率往往较低。这些独特的反应特性以及令人惊讶的生物活性激发了研究人员设计不同类别的含硒化合物用于癌症治疗。近几十年来,合成了多组有机硒化合物,并对其抗肿瘤活性进行了深入研究。
在所有合成特征和硒有机化合物,其中一些已被证明是有效的对几种类型的癌症细胞系,如结肠癌细胞系(HT29),乳腺腺癌细胞系(mda-mb-231),胰脏癌细胞系(PANC-1),恶性黑色素瘤细胞系(UACC903)(图1)[3]。然而,这些化合物的抗癌作用机制通常仍不清楚。根据文献,硒可能通过两种不同的途径抑制癌细胞的生长。在正常细胞中,硒可以作为一种抗氧化剂帮助这些细胞抵御氧化损伤,而在肿瘤前细胞中,硒会转化为一种预氧化剂,使这些细胞更容易受到硒的攻击。因此,据报道,肿瘤细胞一般更容易受到硒化合物所表现出的细胞毒性作用的影响。此外,有机硒化合物的抗癌作用还可通过细胞凋亡、抑制血管生成、调节血管活性等途径实现。基于这些发现,考虑将硒与化疗或化学预防药物结合,比如SAHA和阿司匹林等,提供新的含硒化合物具有更强的或更好的抗肿瘤药效果被认为是一种很有前途的方法选择性识别新的抗癌药物类别(图2)。
迄今为止,已报道了几种结构特征各异的有机硒化合物。目前对这些化合物的研究主要集中在其生物活性方面[20-22]。从有机合成的角度,我们认为,系统地总结合成具有强大抗癌性能的有机硒化合物的方法,对于进一步研究发现新的含硒肿瘤治疗衍生物也具有重要意义。本文从含硒有机分子的结构特点出发,对近年来报道的含硒有机分子进行了分类,并对其合成方法进行了综述,对今后的新型含硒生物活性小分子的设计、合成及结构修饰的研究具有一定的指导意义。
2.不同类型有机硒化合物的合成方法
2.1 硒氰酸酯和硒异氰酸酯
硒氰酸酯和硒异氰酸酯在肿瘤中对多种信号通路具有抑制作用。甘瑟首次报道了将硒氰酸盐引入有机化合物,硒氰酸钾(KSeCN)作为亲核试剂与1,4-二溴甲基苯(8)的偶联反应得到1,4-双(硒氰基-甲基)苯(9)(p-XSC),它是一种强化学预防药理含硒化合物(方案1)。此后不久,Ghose对人口腔癌进行p-XSC的检测,发现其化学预防作用是由丝裂原激活蛋白激酶和Fas通路诱导的。
硒氰酸酯与现有抗癌药物最著名和最成功的组合之一可能是SAHA的结构修饰,SAHA是一种现有的组蛋白去乙酰化酶抑制剂。硒氰酸盐与SAHA的结合显著增加了对组蛋白去乙酰酶的抑制活性(方案2)。含硒阿司匹林衍生物7,其IC 50值达到2.1-3.4 nM,是氟脲嘧啶(5-FU)治疗结肠癌阳性对照(方案2)[19]的10倍。此外,它对正常细胞的细胞毒性也降低了,表明增强了安全性。Roy和他的同事发现2-(5-溴戊基)-7-氨基苯并(de)索奎诺林-1,3-二酮(14)对转化细胞具有潜在的抗血管生成和细胞毒性作用。此外,他们也试图将硒氰酸酯部分移植到该结构上,以进一步提高生物活性(方案2)。对合成的硒氰酸酯衍生物(16)的机制研究表明,其抗癌作用可能是通过p53依赖性凋亡诱导的。对于硒异氰酸酯的构建,传统的策略是霍夫曼碳胺反应。KOH和三氯甲烷处理初级胺时,原位生成卡宾结构,在回流的THF中,硒和三乙胺反应生成相应的硒异氰酸酯。也有报道称,光气可用于生成异氰酸酯,并可进一步转化为硒异氰酸酯(方案3)。
Fernandez-Bolanos报道了一种一锅法合成硒异氰酸酯的方法。苯烷胺类甲酰化后,相应的甲酰胺经过三光气和硒的反应,得到相应的硒异氰酸酯的衍生物19和27 - 29(方案3)。该系列化合物的生物活性评价表明, 硒异氰酸酯可以抑制黑色素瘤细胞生长,前列腺癌和乳腺癌等IC 50值达到10-40μM,如表1所示,稍微高于相应的异硫氰酸酯,这些异硫氰酸酯的IC50值从20~50μM 不等。这种一锅法的策略是很好的,由于气相光气可以被三光气所取代,因此传统的气相光气合成方法难以实现。此外,硒异氰酸酯可以以甲酰胺为原料一步合成。
2.2 硒化物
硒化物结构存在于许多含硒氨基酸衍生物中,如硒代蛋氨酸(Se-Met, 32)、硒二谷胱甘肽(SeGSH)和甲基硒半胱氨酸(35),目前被广泛认为是硒在哺乳动物中可能的存在形式(方案4)。然而,据我们所知,SeGSH的合成尚未见报道。这些含硒氨基酸在体内外均经过检测,对不同的癌细胞系如人乳腺癌细胞、人结直肠癌细胞等均表现出较强的抑制作用。由于该类分子通常具有手性中心,因此构建含硒氨基酸的合成方法有两种。一种方法以纯对映体为原料。例如,Cao从d -蛋氨酸(30)经几个步骤合成了31。甲烷硒酸锂被用来将单硒官能团引入31(方案4)。
另一种方法是将含硒氨基酸作为外消旋体混合物,通过水解酶进行拆分,具有较高的手性选择性。例如,Liu报道了一种合成L-甲基硒半胱氨酸的方法(35)。首先通过33与甲硒酸钠(NaSeMe)反应合成了一对硒半胱氨酸对映体。采用L-氨基酰基酶催化水解,分离出两种对映体(方案4)[41]。
Kang报道了苯氨基乙基硒化物39和40在体内和体外对前列腺癌均有抑制作用(方案5)[42]。该反应的机理可能与二硒酰胺的还原有关。原位生成的还原硒可以作为亲核试剂进攻37生成目标化合物39和40。
铂(II)顺铂、卡铂、奥沙利铂等配合物药物在癌症治疗中应用广泛。由于与Pt(II)配合物的结构相似,Au(III)配合物在这方面也受到了广泛的关注。有报道称,与顺铂相比,部分Au(III)配合物对不同的癌细胞株具有更强的抑制作用,说明Au(III)配合物也可以作为一种新型的抗癌药物系列[43]。Ruan在前人研究的Au(III)衍生物43的基础上,设计合成了一种硒修饰的卟啉Au(III)复合物42[44]。机理研究表明,42通过诱导线粒体依赖的凋亡和抑制细胞周期而表现出较强的细胞毒性(方案6),硒修饰的卟啉Au(III)复合物的细胞毒性作用强于原Au(III)复合物(43)和顺铂,如表2所示。
甲基硒取代基也被引入核苷结构。林报道2 ',3 ' - 5 ' –Se修饰D一核糖衍生物(46,49和51)能够诱导半胱天冬酶的细胞凋亡在不同肿瘤细胞系(表3)。在他们的合成策略中, 化合物44的2,2’分子内环合是在碳酸二苯酯和碳酸氢钠存在的条件下,在DMF中反应得到2,2’-无水尿苷 (45),然后再经过二甲基联硒化物和硼氢化钠的处理得到46。该策略对3 ' -甲基胸苷(49)的合成也很有效。然而,在2,3 ' -分子内环化之前,需要通过甲基化反应激活3 ' -羟基。48的后续开环反应在相似的反应条件下进行,但温度较高。林将这种现象归因于环应变较小,与45相比。对甲苯磺酰酯别选择性的引入到化合物50中5′c位置作为活性离去基,构建5′-甲基硒尿苷。然后,在硼氢化钠存在的条件下,所得到的甲苯磺酸酯与二甲基二硒发生偶联反应,形成单硒酰胺(Scheme 7)。
Greenberg及其同事报道,在碱基片段引入硒也可以实现核苷的硒修饰。在这种情况下,糖基上的羟基应首先得到充分保护。52经过NBS的介导提供烯丙基溴,然后用二苯基二硒酰胺和硼氢化钠处理,得到单硒酰胺(54),过程与上面讨论的类似(图8)。
芳硒的合成方法通常涉及重氮盐的形成。例如,最近设计合成了一系列甲基硒与异共促食素和苯他汀衍生物的连接物。在这项工作中,作者打算用甲基硒取代天然产物isoCA-4中的C-3、C-4和C-4’的甲氧基,发现C-4的取代几乎不可能发生。根据他们的解释,两个邻位甲氧基可能抑制反应活性。因此,他们试图将甲基硒连接到C-4’位置,尽管结果显示衍生物的细胞毒性并不比异辛卡-4高。基于这些发现,他们主要关注C-3取代。本系列中活性最强的分子61a对不同的癌细胞株具有抑制作用,其IC50值为2-11 nM。(表4)力学研究表明,61a具有管蛋白的功能,就像isoCA-4一样。在合成过程中,重氮盐作为离去基引入硒氰酸盐基团。合成的硒氰酸酯(57)经硼氢化钠还原,随后与碘甲烷进行甲基化,得到甲基(芳基)硒片段。(方案9)Wen报道了一系列含硒/硫的a-4类似物,并检测了它们对SGC7901、HT1080和KB细胞株的生物活性,见表5[48]。用硒取代吲哚结构取代了CA-4的B环和顺式双键。以1,2-双(3,4,5-三甲氧基苯基)二硅烷(64)为硒源,通过重氮盐63与二硅烷钠的偶联反应得到。在三氯化铁和碘的存在下,二硒酰胺键断裂,在微波反应下把吲哚基部分连接起来。(表10)。
Moreno报道了一系列含硒嘧啶衍生物[2,3-d],在体外显示了强大的细胞毒活性(表6,方案11)[49]。与一般构建硒化芳基的方法不同,他们通过硒异氰酸酯和邻氨基芳腈的环化或用硒脲亲核取代氯基,成功构建了含硒嘧啶[2,3-d]。硒元素的引入不需要重氮盐的形成。在生物活性研究中,他们还测试了其中间体硒醇68,73和74的细胞毒活性并发现68a (X=C)也具有较强的细胞毒活性。
2.3 联硒化物
取代二硒醚类化合物由于在小鼠肝脏、肾脏和大脑中易于分布,已在生物系统中显示出有趣的药代动力学特性。因此,取代二硒醚类化合物有潜力开发成为有前途的抗肿瘤药物脑癌或肝癌。Battin公布环硒二聚体(2),拥有强大的对不同细胞DNA损伤抑制IC 50值变化从3.34微摩尔到25.1微摩尔 (Scheme12) [51]。
对于有机硒分子中二硒酰胺结构的合成,通常采用Na2Se2或Se3CN2作为硒源[51-56]。与单硒类化合物的生成不同,单硒类化合物的生成通常涉及到小二硒类化合物的原位还原,二硒类化合物的合成可以通过直接亲核攻击实现,从二硒类化合物(如Na2Se2或Se3CN2)到另一个带有活性离去基的偶联(方案13-15)。
采用类似的策略,通过Na2Se2或[Se+ N2H4]在芳香环上取代活性氯来构建芳香二硒醚类化合物(方案13-14)。黄合成的1,2-二(喹唑啉-4-酰基)二硒醚(77)对不同的癌细胞如A549、PC-3、MDA-MB-435等具有广谱的抗癌作用。
由金合成的一系列1,2-二(吡啶-3-酰基)二硒醚衍生物(79a-j)具有对MCF-7的抗多聚性活性,如表7所示。根据文献,有几种不同的合成策略来构建对称二硒醚类化合物(方案15)。例如,用苯基氨基与作为硒源的Se3CN2反应得到中间产物硒氰酸酯衍生物(80)。随后,对关键中间体80进行硼氢化钠的还原,得到了理想的二硒酰胺化合物[54,56]。谷口教授开发的另一种策略依赖于芳基碘化物(82)与硒在铝、2,2'-双吡啶和碘化亚铜[55]存在下的直接耦合反应。虽然这种方法一般进展缓慢,但可以避免使用硼氢化钠等还原剂。
2.4 亚硒酯类
最近,郭和他的同事将硒作为硒酯引入碳水化合物中。用二氯化亚硒酸盐处理不同糖时,观察了区域选择性酯化反应。对于蔗糖,可以得到单酯(85)。当二氯化亚硒酸盐应用于木糖醇时,得到了五元环亚硒酸酯(87)(方案16)。85和87对不同的癌细胞株均有较好的抑制作用。
其中最常用的无机硒源之一亚硒酸钠已被用作营养补充剂。近年来,亚硒酸钠也被用于治疗癌症。但有报道指出,亚硒酸钠的毒性作用不容忽视。亚硒酸钠的进一步研究表明,Na2SeO 3在极低浓度下对癌细胞(SMMC-7221)和正常细胞(HL-7702)均表现出明显的细胞毒性, 85和87可选择性抑制SMMC-7221在0.15 ~ 1.2 ppm Se的检测浓度下,表现出较高的选择性[57,58]。
2.5 取代的硒脲
取代硒通常被引入抗氧化分子中,以进一步增强其生物活性[60-62]。据报道,含硒脲结构的化合物可作为活性氧(ROS)的清除剂,是一种很有前途的抗癌策略。Calcatierra受席夫碱衍生物triapine(92)的启发,报道了一系列硒脲衍生物(91)具有抗氧化、清除活性氧等多种生物活性,如表8[60]所示。Laura将硒脲与一些内源性的生物活性甾体结构连接起来,在进一步的生物活性测试中,结果显示含硒衍生物(94)清除活性氧增加,对癌细胞具有协同作用[61](方案17,18)。
Plano报道了一系列对PC-3细胞系具有较强细胞毒性作用的双嘧磺酰氨基甲酸盐(BSC)衍生物,见表9[63]。本系列中对PC-3最有效的衍生物96n的作用机制研究表明,BSCs治疗肿瘤细胞的凋亡可能是由致死的有丝分裂诱导的。与上述硒脲类衍生物不同,BSCs的合成往往是从各种烷基氨基咪唑烷酸氢碘化物开始的(95)和不同的酰氯,在吡啶作为催化剂的存在下,在室温下以1:2.1摩尔的比例反应(方案19)。
文献表明,二苯基取代硫脲是一种强的碳酸酐酶抑制剂,可在低氧条件下阻断肿瘤细胞的生长[64]。最近,Fabrizio Carta及其同事报道了一系列取代硒脲类化合物在常氧和低氧条件下对hCA亚型的抑制作用以及对PC-3、MDA-MB-231和HT-29的活性(Scheme 20)[65]。在PC-3和MDA-MB-231, 98a和98b中,在常氧条件下表现出与对照化合物相同或更强的抑制作用。三种化合物对HT-29均无活性(图4)。
然而,所得的衍生物对不同类型的CA异构体没有很好的选择性。由于CA I和CA II广泛分布于健康细胞中,这些衍生物可能对正常细胞产生影响。因此,可能需要对这些含硒化合物进行进一步的安全性研究。
2.6硒代亚砜
硒氧化二甲酯(101)是一种最简单的含硒生物活性分子,对人前列腺癌细胞株(LNCaP)具有较强的细胞毒作用[66]。随后,对其他癌细胞株的抗癌活性进行了测试。例如,二甲基氧化硒进一步检测对乳腺癌细胞(mda - mb - 231)与IC 50值为0.105μM [67]。治疗后的二甲基硒氧化物,即使在低剂量,两种癌症细胞活力下降[68]。硒化二甲酯的形成是通过m-CPBA或过氧化氢介导的二甲基硒过氧化反应完成的(方案21)[69]。
通过对单硒烯类化合物的制备和生物评价,文进一步合成了其硒氧化物类似物[48]。根据他们的方法,过氧化氢被进一步用于硒化物衍生物的氧化,得到硒氧化物衍生物(方案22)。一般来说,含硒氧化物连接剂的化合物表现出较弱的抑制作用,除了102a。(表10)
2.7含硒的杂环化合物
由于杂环结构的多样性,硒元素的引入可以提供不同的含硒杂环,具有不同的生物活性。含硒杂环化合物的合成策略因杂环结构的不同而异。
2.7.1 硒恶唑烷酮衍生物
临床试验中第一个硒唑烷酮衍生物是依布硒林 (3),它首先被用作抗氧化剂,并被用于治疗噪音引起的听力损失。对晚期癌症化疗致听力损失的防治也进行了的研究。另一个衍生物,1、2 - (双 (1, 2-苯硒唑烷-3 (2 氢)酮))乙烷,名为乙烷硒啉(103)随后被设计合成,作为一种治疗胃癌、肺癌和结肠癌的有效的硫氧还蛋白还原酶抑制剂。Li报道了一系列硒唑烷酮衍生物(103-106)作为强效硫氧还蛋白还原酶抑制剂,有望进一步开发成为新型抗癌药物(图5)[70]。
构建硒唑烷酮结构的关键中间体为2-(氯代羰基)苯基次氯酸盐(109),用氯化亚砜对2,2'-异丙二基二苯甲酸进行处理可得到该结构。2-(氯代羰基)苯基次氯酸盐(109)与不同胺的偶联反应提供了多种具有硒唑烷酮结构的依布硒啉衍生物,其产率极好(图23)[12,71]。
最近,Sucheck和他们的同事开发了一种针对依布硒啉及其衍生物的新方法[72]。他们描述了硒氰酸钾(KSeCN)与N-取代的邻卤代烷之间的一种新的光热诱导铜介导的交叉偶联反应,形成2-烷基-1,2-苯并异硒噻唑-3(2H)-酮(Scheme 24)。有趣的是,在加热过程中,铜配体(1,10-菲罗啉)通过原子转移(AT)机制促进了C - Se键的形成。另一方面,在没有配体的情况下,光诱导活化可能通过单一的电子转移(SET)机制进行。根据他们提出的反应机理,AT涉及卤化物原子从芳基卤化物转移到a (phen)CuI (SeCN)络合物,形成笼状芳基(Ar·)和(phen)Cu II (SeCN)X络合物。进一步的偶联反应得到了芳基硒氰酸酯和(phen) cuIx,它可以通过配体交换再生(phen) cuI (SeCN)来完成这个循环。
芳基硒氰酸酯可以环化形成依布硒啉衍生物。一套光诱导的机制需要激进的亲核芳香取代反应(S RN1)和涉及光激发(Cu(SeCN)2)离子—提供一种激发态的(Cu(SeCN)2)−•自由基,可以形成一对激发态的[Ar-X•和CuII (SeCN)2]。随后自由基对的偶联过程提供了芳基硒氰酸酯和[Cu I (SeCN)X]−,它们可以进一步进行配体交换来完成催化循环。
2.7.2. 硒唑衍生物
Srivastava首次报道了一种1,3-硒唑衍生物(117),在体外对P388和L1210细胞具有新的细胞毒性,对小鼠Lewis肺癌具有抗肿瘤活性[73]。硒唑环是由氰化物(113)和3-溴-2-草酸乙酯(114)在Al2 Se3作为硒源的存在下耦合反应而成(方案25)。
2015年,Zhou报道了一系列含有1,3-硒唑骨架的生物活性分子(119),经体内实验证明,这些分子对人乳腺癌MCF-7细胞有效(表11)[74]。进一步的机制研究表明,119g可通过减少ROS的生成诱导MCF-7细胞凋亡。硒异辛酸盐(118)是根据2.1节讨论的合成方法首次制备的,与2-氯吡啶-3-胺偶联得到硒唑衍生物119a-g(方案26)。
2.7.3. 硒酚衍生物
Juang报道一种包含硒酚的新型α三重杂环化合物(1),表现出对几种人类癌症细胞系有抑制活性与IC 50值的范围[75]。该化合物是以硒酚为原料合成的。硒酚首先用琥珀酰二氯(121)或N,N-二甲氧基-N,N-二甲基琥珀酰胺(122)处理,得到1,4-二酮衍生物(123)。随后1,4-二酮衍生物的PaalKnorr环化反应生成吡咯衍生物(124),然后进行Vilsmeier-Haack反应,在吡咯环上引入醛基。用n-BuLi和DMF对124进行处理,在硒吩基上又附加了两个醛结构。最后,硼氢化钠介导的还原反应将三个醛基转化为甲醇基(方案27)。
2014年,Arsenyan报道一系列硒酚 [3、2 c]和[2,3 c]喹诺酮衍生物, 对几种细胞系具有强烈的细胞毒性和IC 50值从6到100μg /毫升不等。不久之后,他们合成了硒苯[3,2-c]和[2,3-c]香豆素衍生物,具有一定的细胞毒性[77]。最近,他们利用这一策略合成了更多的类似物(128a-f, 132a-f),并进行了进一步的生物学研究。[78]机理研究表明,该系列化合物能够抑制特定的抗氧化酶系统,如SOD、GPx(选择性抑制肿瘤细胞GPx活性)和TrxR;降低ROS浓度。在他们的合成过程中,建立了一个共轭烯烃-炔形成了硒酚骨架。随后,原位生成SeBr4(由二氧化硒和浓氢溴酸制备)引发分子内环化,得到了目标硒酚衍生物。
2.7.4. 硒二唑衍生物
有报道称1,2,5-呋喃嘧啶是小分子表达抗癌活性的重要结构基序[31,79 -81]。1,2,5-硒代二唑可视为硒代氧氟杂氮。因此,设计并合成了多种含硒二唑骨架的分子。他们的生物活性测试得到了令人兴奋的结果。例如,Liang合成并评价了一系列硒二唑衍生物(135-137),它们可以诱导不同癌细胞株caspase和p53依赖的凋亡。肿瘤细胞在x射线照射下(8 Gy)细胞毒作用显著增强,说明该系列化合物可增强肿瘤细胞的放射敏感性(图6)[80]。
构建苯并苯二唑骨架的一般步骤是邻二胺与SeO2的偶联反应(方案29-31)。结果表明,微波辅助反应可以使这种转化更加有效。并且可以在较短的反应时间内得到目标硒代唑,收率较高(方案30)。张和他们的同事设计并合成了一系列含硒二氮唑的CA-4衍生物。构建硒二唑环可以在结构中引入硒元素,在一定的时间内,还可以构造出有机硒CA-4衍生物的构象。在本系列中,143a/b化合物中有两种具有比CA-4更强的细胞毒活性,如表13所示。在本例中,除邻二胺外的邻二酮肟(142)被用作偶联反应物之一(方案31)。
Arsenyan报道了一系列具有不同酰胺取代基的1,2,3-硒二唑衍生物[82]。进一步对这些化合物在HT-1080(人)和MG-22A(小鼠)上的体内生物学评价表明,这些化合物具有强大的抗肿瘤活性。1,2,3-硒二唑的形成与乙基乙酰乙酸氨基脲(144)与二氧化硒的环化反应有关。然后在室温将所得酯(145)转化为不同的酰胺,得到化合物146-149(方案32)。这些反应均在黑暗中进行,以防止硒二唑循环光降解。
2.7.5. 四氢化硒酚衍生物
由于硒和氧的性质相似,核苷酸衍生物的一种实用的结构修饰策略是用硒原子取代糖骨架上的氧原子。按照这一策略,Kim报道了一系列含硒核苷酸[83]。其中一个最强大的衍生品153年可能抑制HCT116、A549, SNU638, T47D和PC-3细胞系在浓度为1微摩尔时。合成方法中关键的环化反应是Na2Se和甲磺酸盐的连续亲核取代。合成的中间产物152经过硒氧化、尿嘧啶连接减少和去保护得到153(方案33)。
2.7.6.硒罗丹明衍生物
Hill报道了一种罗丹明衍生物(159),可作为光敏剂用于表达p- gp的癌细胞的光动力学治疗[84]。这种硒罗丹明衍生物提供了一个固氮原子与黄嘌呤核结合的理想光谱值,可以有效地对抗p- gp表达细胞。154和155的偶联反应在s-BuLi和TMEDA存在下进行。在乙腈中,在POCl3 /Et3N存在下,硒化二芳基(156)进行了后续环合反应。最后,在HPF6中,化合物157和158的偶联反应得到了硒罗丹明衍生物(159)(Scheme 34)。
结论
迄今为止,含硒分子的设计通常遵循两种不同的策略。其中一种采用了生物同分异构的概念,即在生物活性化合物中以氧或硫原子取代硒原子。另一种方法是以硒化物、硒(异)氰酸盐的形式引入硒基,取代硒脲或硒酯来报道生物活性分子或批准的抗癌药物,目的是进一步增强其生物活性或提高细胞毒性选择性。
一般来说,由于不同含硒分子结构的差异,构建不同含硒分子的合成方法也不尽相同。硒源既可以作为亲电试剂,如元素硒,也可以作为二烯类化合物或亲核试剂,如硒氰酸盐或甲烷烯醇酸盐。对于含硒杂环,硒源对环化反应的参与程度因杂环结构骨架的不同而异。本文综述了近年来含硒抗癌药物的合成方法,希望对今后新型含硒抗癌药物的设计和合成有一定的指导意义。
本文由福山生物整理翻译,转载请明确注明出处。
上一篇:硒的预防癌症