






最新资讯



-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
Nrf2:阿尔茨海默氏病治疗中的黑马
发表于:2021-09-01 作者:admin 来源:本站 点击量:30831
原文:Osama A , Zhang J , Yao J , et al. Nrf2: a dark horse in Alzheimer's disease treatment[J]. Ageing Research Reviews, 2020, 64.
翻译:
阿尔茨海默氏病(AD)是一种年龄依赖性神经退行性疾病,是痴呆症的主要原因。AD的常见特征包括淀粉样β肽(Aβ)聚集,高水平的高磷酸化tau蛋白(p-tau)和氧化还原稳态失败。迄今为止,所有针对Aβ和/或p-tau的药物临床试验均失败了。在AD大脑中已经观察到转录因子Nrf2(核因子-类胡萝卜素2-p45衍生因子2)及其驱动基因(NQO1,HO-1和GCLC)的表达下降,以及Nrf2相关途径的改变。Nrf2在维持细胞氧化还原稳态和调节炎症反应中起关键作用。Nrf2激活还提供针对日益增加的病理(包括神经退行性疾病)的细胞保护作用。这些证据表明,Nrf2激活可能是一种新颖的AD治疗选择。有趣的是,最近的研究还表明,Nrf2会干扰AD中的几种关键致病过程,包括Aβ和p-tau途径。本综述旨在深入了解Nrf2在AD中的作用。此外,我们还讨论了有关Nrf2激活剂用于AD治疗的进展和挑战。
关键词:阿尔茨海默病、Nrf2、 Keap1、β淀粉样蛋白肽、Tau蛋白、氧化应激
1. 前言
神经元主要参与学习、思考、记忆和计划(Carter等人,2019)。神经退行性疾病的主要原因是多种病理因素导致神经元功能和生存能力的逐步下降。这些多因素神经退行性疾病正在影响全球数百万人(Calderon-Garciduenas和Duyckaerts,2017)。神经退行性疾病,例如阿尔茨海默氏病(AD),被证明与蛋白质折叠错误、线粒体损伤和氧化应激(OS)有关。AD是一种不可逆的进行性脑部疾病,会逐渐发生并导致记忆力减退以及思维和行为下降(Lu等人,2018年; Ossenkoppele等人,2015年)。AD被认为是痴呆症的主要原因,这是至少两个认知领域的逐步衰退,并导致正常的社交或职业活动残疾。AD通常会影响老年人,但它不是正常的衰老缺陷。AD的特征包括淀粉样β肽(Aβ)的聚集、高磷酸化tau蛋白(p-tau)的积累、炎性介质的产生、OS以及胆碱能、突触和认知功能的障碍。遗传、环境因素和一般生活方式是与AD相关的病因因素的一部分(Long和Holtzman,2019)。当前,AD影响着超过5000万人,估计2050年全球发病率将达到1.5亿(Jannat等人,2019)。由于仅部分了解AD的发病机理,因此AD的治疗仍然是一个挑战(Park等人,2018)。许多III期临床试验都失败了,尤其是针对Aβ产生和聚集的试验。由于缺乏明显的疗效,最近终止了Crenezumab(NCT03491150)、lanabecestat(NCT02972658)和solanezumab(NCT01900665)临床试验。在寻找治疗AD的有效药物方面的系统性失败,促使科学家们探索新的策略来对抗这种疾病。
AD具有多种病理特征,例如大量的DNA氧化、严重的线粒体损伤、广泛的脂质过氧化作用、高水平的神经毒性微量元素以及Aβ的升高(Deibel等,1996; Huang等,1999; Wang等(2005年)。所有这些因素都会增加自由基或活性氧(ROS)的形成,因此,因此AD患者的大脑也受到OS的影响(图1)。在生理条件下,ROS的动态平衡受到ROS生成系统和细胞抗氧化网络的良好调控(Zhang等,2020; Zhang等,2019)。抗氧化防御系统包括几种酶和小分子,例如血红素加氧酶-1(HO-1)、超氧化物歧化酶(SOD)酶、谷胱甘肽(GSH)、过氧化氢酶(CAT)、硫氧还蛋白(Trx)、硫氧还蛋白还原酶(TrxR)、谷胱甘肽过氧化物酶(Gpx)家族和NAD(P)H:醌氧化还原酶1(NQO1)。在人类AD研究中,患者的大脑显示出核因子红系2相关因子2(Nrf2)、SOD1、CAT和Gpx的水平降低(Chenere等,2007; Mota等,2015)。由于Nrf2和这些重要的抗氧化酶含量低,AD大脑容易受到ROS的侵害。在这种情况下,ROS对大脑造成进行性和不可逆的损害。用抗氧化剂(例如维生素E、维生素C和雌激素)直接治疗可能在AD中发挥积极作用,但这些抗氧化剂在临床患者中的疗效仍处于初级阶段(Feng和Wang,2012)。通过Nrf2活化来增加抗氧化蛋白的水平已被认为是实现神经保护的更好方法(Buendia等人,2016; Chen和Maltagliati,2018)。 Nrf2诱导大约250个基因编码不同的细胞保护和解毒蛋白的表达。高水平的Nrf2可以缓解AD中ROS和/或线粒体功能障碍引起的损害(Eftekharzadeh等人,2010; Fu等人,2018)。最近的研究表明Nrf2可以预防AD中某些关键的早期致病过程,包括Aβ(Cuadrado等,2018; Han等,2019)。已知Nrf2可通过调节抗氧化蛋白和解毒蛋白发挥神经保护作用,但是Nrf2具有干扰β-分泌酶(BACE1)、Aβ和p-tau的能力是一个有趣的发现。该领域的一些优秀的研究是由不同的研究小组进行的(Bahn等人,2019; Kitaoka等人,2019; Sotolongo等人,2020; Tang等人,2018),在此,我们旨在回顾和讨论这些发现,以提高我们对Nrf2在AD中的作用的认识。理解Nrf2激活剂针对Aβ和p-tau的神经保护作用的潜在机制可能有助于新AD药物的设计和开发。此外,我们讨论了Nrf2激活剂作为AD潜在疗法的进展以及开发Nrf2激活剂的主要挑战。在药物设计过程中考虑这些问题可能会增加临床试验的成功率。
2. Nrf2的结构和功能
Nrf2蛋白是NFE2L2基因的产物,是研究最多的Cap 'n 'collar基区亮氨酸拉链(b-Zip)转录因子家族成员之一。Nrf2自1994年被发现以来,关于该转录因子的研究不断增多。学术界和产业界之所以对Nrf2如此感兴趣,很大程度上是因为Nrf2具有广泛的功能。Nrf2是细胞保护系统的主要调控器,即II相解毒酶和抗氧化剂 (Cuadrado et al., 2019)。Nrf2参与NADPH、NQO1、HO-1、胆绿素还原酶、谷胱甘肽家族和硫氧还蛋白系统的转录调控。基于Nrf2的转录程序的细胞保护作用使细胞适应并在应激条件下生存(Hou et al., 2018)。这些功能会改变代谢和生物能,从而影响中间代谢和线粒体功能(Kitaoka et al., 2019; Merry and Ristow, 2016)。由于Nrf2调控多种抗氧化酶的表达,其抗氧化作用已被广泛研究(Peng et al., 2015b)。Nrf2在自噬调控中的作用已经被许多实验室解决,他们观察到Nrf2激活导致自噬增加,而Nrf2缺乏导致自噬减少(Frias et al., 2020; Jo et al., 2014; Komatsu et al., 2010; Pajares et al., 2018)。激活自噬可增加溶酶体介导的蛋白降解和损伤细胞器及感染微生物的清除(Tang et al., 2018)。Nrf2还参与调控另一种类型的细胞死亡,即铁依赖性脂质过氧化引起的铁死亡。Fenton反应(图1c)可能部分解释了铁死亡对铁的依赖,因为具有氧化还原活性的铁池可以催化脂质过氧化,形成损伤,最终导致细胞死亡。与其他类型的细胞死亡相比,铁死亡表现出独特的形态、生化和遗传特征(Dixon et al., 2012)。由于脂质过氧化是铁死亡的核心特征,脂质过氧化物清除剂能有效中和铁作用(Homma et al., 2019; Matsushita et al., 2015)。有趣的是,到目前为止,所有涉及铁蛋白作用的基因都受到Nrf2的转录调控,包括但不限于Gpx4、Trx、TrxR、HO-1、铁转运蛋白和铁螯合酶。因此,Nrf2可以作为这一过程的主要调控因子。人们对探索铁死亡在包括AD在内的神经退行性疾病中的作用越来越感兴趣(Cong et al., 2019; Do Van et al., 2016)。然而,还需要进一步的研究来验证铁蛋白增多与神经退行性疾病的联系。Nrf2激活的另一个显著特征是炎症抑制。Nrf2的抗炎能力比预期的更为复杂, 它涉及Nrf2靶基因编码蛋白的转录上调(如白三烯B4脱氢酶) 以及编码的主要促炎细胞因子(如IL-6和IL-1b)基因的下调 (Dick et al., 2001; Kobayashi et al., 2016)。OS和炎症是包括癌症、心血管和神经退行性疾病在内的大多数人类病理的主要因素(Kwak and Kensler, 2010; Liby et al., 2012)。特别是Nrf2在神经退行性疾病、糖尿病和肾脏疾病中的作用是目前研究的热点 (Das et al., 2013; Fao et al., 2019; Matzinger et al., 2018; Yamawaki et al., 2018)。
不幸的是,到目前为止,Nrf2结构还没有被完全阐明。人类Nrf2蛋白由605个氨基酸组成。由七个区域组成,命名为Neh1-Neh7 (Nrf2-ECH同源性)。Neh1、Neh3和Neh6位于c端。另一方面(n -端)包含Neh2、Neh4和Neh5结构域。Neh7是由Wang等人发现的,与Neh5相邻。Neh1负责DNA结合(图2a) (Wang et al., 2013)。Neh2通过两个基序DLG和ETGE与kelch样ech相关蛋白1 (Keap1)相互作用(Nioi et al. , 2005)。Neh3是一个反激活区域,可以提高抗氧化反应元件(ARE)依赖基因的转录(McMahon et al. , 2004)。Nrf2的keap1独立降解主要由Neh6区域介导。Neh7可能与视黄酸受体(RAR α)结合,这种相互作用导致Nrf2-ARE信号通路的负调控(Wang et al. , 2013)。
3. Nrf2调控的分子机制
Nrf2调控大量基因的表达。同时,它也是大量生物分子的靶点。这些生物分子以不同的机制调控Nrf2功能。了解Nrf2的调控机制对于开发靶向Nrf2的新分子以及了解Nrf2的治疗作用具有重要意义。Kwak等人描述了Nrf2通过结合其启动子区域激活其表达的能力,这支持了Nrf2自我调节(autoregulation)的观点(Kwak et al., 2002; Miao et al., 2005; Rushmore et al., 1990)。Nrf2的调控除了自身调控外,主要分为keap1依赖性和非keap1依赖性调控。这里我们总结了Nrf2调控的机制(图3)。
3.1. Keap1依赖性调控
自从Itoh等人发现去除Nrf2的Neh2可以增加Nrf2活性以来,Keap1在Nrf2调控中的作用就已经为人所知(Itoh et al., 1999)。人Keap1(基因ID: 9817)蛋白是由624个氨基酸残基组成的富含半胱氨酸的蛋白,表观分子量为69.7 kDa (Itoh et al., 1999; Zipper and Mulcahy, 2002)。Keap1有5个具有不同功能的结构域,即N端区域(NTR;1-60),C末端区域CTR(599-624)、BTB(61-179)(Broad complex, Tramtrack and Bric a Brac)/POZ(poxvirus and zinc finger)、双甘氨酸重复区DGR (315-598) (double glycine repeat or Kelch repeats)及插入区IVR(180-314)(intervening region)。DGR和CTR一起被称为Keap1-DC,它对Nrf2的Neh2结构域的结合至关重要。人Keap1含有27个半胱氨酸(Cys)残基,但只有Cys151、Cys226、Cys273、Cys288、Cys434和Cys613对Nrf2激活至关重要(图2b) (Bryan et al., 2013; Canning et al., 2013; Canning et al., 2015)。
Nrf2是Keap1最具特征的靶蛋白之一。在基础条件下,Keap1有利于Nrf2蛋白酶体降解,以防止ARE的组成性激活(Huang et al., 2000)。人们提出了不同的模型来理解这种相互作用和调节的机制。铰链和插销模型是理解Keap1-Nrf2相互作用的首选模型(图4)。在这个模型中,Nrf2 ETGE基序首先与Keap1结合。ETGE结构域采用圆转构象,深深插入Keap1口袋,埋藏面积为420 Å2 (Abiko et al., 2011; Wakabayashi et al., 2004)。然后Nrf2通过其DLG基序在Keap1的第二个独立结合位点结合。通过对ETGE和DLG的比较,可以看出两者都与Kelch域口袋的底部相互作用,但其构象和亲和力不同。热力学和动力学分析表明,与Keap1-DLG较弱的相互作用相反,Keap1-ETGE是焓驱动的,开关速率较慢,导致高亲和力的相互作用(Hong et al., 2005; McMahon et al., 2003; Tong et al., 2007)。在激活模式下,DLG基序与Keap1分离,导致Nrf2的释放和转位到细胞核(Bloom and Jaiswal, 2003)。
Nrf2的另一个keap1依赖性调控是通过p62。p62蛋白也被称为死骨片1 (SQSTM1)。p62蛋白可以结合多聚泛素化蛋白和自噬机制,靶向自噬的特定载体(Kim et al., 2008; Pankiv et al., 2007)。Komatsu等证明p62通过与Keap1的相互作用破坏Keap1-Nrf2系统(Komatsu et al., 2010)。p62通过其与Nrf2 ETGE相似的STGE基序与Keap1结合。p62高表达导致Nrf2水平升高,反之亦然(Copple et al., 2010)。另一项研究也表明,p62的积累导致Nrf2的一致激活(Inami et al., 2011)。为了理解p62-Keap1结合模型,人们做了很多有价值的工作,结果证明该模型与Keap1的CTR重叠(Komatsu et al., 2010;Lau et al., 2010)。基于编码p62蛋白的mRNA的存在,缺失了与Keap1结合所必需的Keap1相互作用区域,Kageyama等人证明,该变异增加了Keap1的数量,并增强了Nrf2的泛素化。本研究表明,p62 pre-mRNA的剪接负调控小鼠Keap1-Nrf2通路(Kageyama et al., 2018)。然而,关于人类的这种变异还需要更多的研究。BRCA2(PALB2)的伴侣和定位剂二肽基肽酶3(DPP3)也通过ETGE基序与Keap1相互作用,激活Nrf2(Hast et al., 2013)。
3.2. 非Keap1依赖性调控
Nrf2中非keap1依赖性调控也被几个研究小组研究过。在这方面,Nrf2的翻译后修饰,包括磷酸化、泛素化和乙酰化得到了很好的表征。Nrf2含有许多苏氨酸、丝氨酸和酪氨酸残基,是潜在的磷酸化位点。几种激酶可能磷酸化Nrf2并调节其活性(Rojo et al., 2012)。蛋白激酶C (PKC)磷酸化Nrf2的Ser40 (Neh2),从而破坏与Keap1的关联,进而促进Nrf2转位进入细胞核(Huang et al., 2002)。最近, Fao等人表明,Nrf2在Ser40被c-src依赖激酶磷酸化激活,在Tyr311通过PKCδ磷酸化 (Fao et al ., 2019)。酪蛋白激酶II (CK2)在Nrf2特异序列中也有大约13个潜在的磷酸化位点(Neh4和Neh5) (Pi et al., 2007)。这些位点的磷酸化与易位进入细胞核的Nrf2相连接。研究发现,在CK2抑制剂存在的情况下,Nrf2易位降低(Patrick L. Apopa et al., 2008)。类似地,蛋白激酶R (PKR)样内质网激酶(PERK)介导了Thr80的磷酸化并有利于Nrf2的激活(Cullinan and Diehl, 2004)。糖原合成酶激酶-3蛋白(GSK-3磷酸化)使Nrf2的Ser335和Ser338位点磷酸化并导致其降解(Rada et al., 2011; Rojo et al., 2008)。GSK-3蛋白还作用于酪氨酸激酶Fyn的上游,可以磷酸化Nrf2中的Tyr568,导致Nrf2的核积累(Jain and Jaiswal, 2006,2007)。几种丝裂原活化蛋白(MAP)激酶也显示了不同Nrf2丝氨酸和苏氨酸残基的磷酸化能力。虽然,c-Jun n末端激酶1 (JNK1)和细胞外调节蛋白激酶2 (ERK2)介导的磷酸化诱导了Nrf2的激活,但p38通过磷酸化Nrf2促进keap1和Nrf2之间的联系,从而阻止其核易位(Keum et al., 2006)。Hrd1 (E3泛素连接酶)又称滑膜泛素连接酶。Hrd1对内质网相关降解(ERAD)至关重要。Wu等证明Hrd1与Nrf2的Neh4-5结构域相互作用导致Nrf2泛素化并降解(Wu et al., 2014)。CBP/p300介导的Nrf2赖氨酸残基乙酰化(Neh3/Neh1)增加了Nrf2的转录功能。另一方面,sirt1介导的去乙酰化降低了Nrf2信号(Kawai et al., 2011)。
核因子-κB (NF-κB)蛋白是一组结构相关转录因子,参与了大量的细胞过程。NF -κB可以抑制相关基因的转录,而几个Nrf2催化剂有能力抑制NF-κB活动(Nair et al ., 2008)。大量证据还表明,Keap1通过与IκB激酶-β(IKKβ)结合而在NF-κB通路的负调控中起作用,并赋予NF-κB泛素化和降解作用(Kim et al., 2010; Lee et al., 2009)。
Bach1是b-Zip转录因子的一部分,与小Maf蛋白形成异源二聚体,并作为Nrf2的诱导抑制因子。Bach1/Maf和Nrf2/Maf竞争性地共享同一结合位点(ARE的顺式元素)。因此,靶基因的抑制或激活可能发生。然而,Bach1的亚细胞定位依赖于Nrf2核输出信号和Crm1, Crm1由氧化还原敏感的细胞质定位信号(CLS)驱动,因此Bach1主要由Nrf2调控(Silva-Palacios et al., 2018)。Poganik等人引入了一种新的直接调控Nrf2 mRNA的转录后模型。他们报告说,HuR和AUF1 ,两个mRNA结合蛋白质,可以通过靶向mRNA 3?utr专门调控Nrf2。他们的结果表明,HuR可以促进Nrf2 mRNA的成熟并促进其核输出,而AUF1则稳定Nrf2 mRNA。他们还发现,Nrf2活性的转录后调控在整个脊椎动物水平上是保守的(Poganik et al., 2019)。这一发现为发现更多遵循类似调控机制的Nrf2调控因子奠定了基础。
几项研究已经鉴定出一些通过将Nrf2的mRNA靶向3´UTR来调节Nrf2表达的MicroRNA。这些Nrf2负调节剂的例子包括但不限于miR28,miR-153,miR-142-5p,miR-27a和miR-144(Narasimhan et al., 2012; Yang et al., 2011)。然而,Nrf2与其靶向miRNA之间的关系是高度复杂的,需要更多的研究来发现其他参与Nrf2调控的miRNA及其调控机制以及这种调控在AD发病机制中的作用。综合来看,参与Nrf2调控的多个因素可以为理解Nrf2参与不同疾病和紊乱的治疗提供线索(Cuadrado et al., 2019)。
4. Nrf2在AD中的作用
Nrf2在机体的不同器官(包括大脑)中表达,表达水平各不相同。代谢器官,特别是那些暴露于外部环境的亲电性和氧化剂的器官(如肺和肠) Nrf2水平较高(Riedl et al., 2009)。对人和小鼠大脑中枢神经系统(CNS)的研究表明Nrf2在神经元、星形胶质细胞和胶质细胞中表达(Cuadrado et al., 2019; Shih et al., 2003)。然而,Nrf2在星形胶质细胞中的表达高于神经元。微阵列分析显示Nrf2激活诱导星形胶质细胞中大量Nrf2靶基因(Lee et al., 2003)。在几种神经退行性实验模型中,星形细胞Nrf2表达具有神经保护作用(Cuadrado et al., 2018; Yu et al., 2019)。Nrf2及其驱动基因(NQO1、HO-1和GCLC)的表达在老年人和AD大脑中均出现下降(Silva-Palacios et al., 2018)。Nrf2通过保护性NFE2L2单倍型的高表达可防止神经退行性变。另一方面,Nrf2表达降低与AD风险增加或早期发病相关(von Otter et al., 2010)。Nrf2介导的抗氧化反应的抑制被发现是导致过早衰老表型的关键因素(Kubben et al., 2016)。 Rojo等比较了Nrf2缺乏、衰老和AD小鼠模型之间的差异。他们发现了Nrf2缺乏和衰老之间的7个共同特征,以及Nrf2缺乏和AD之间的10个共同特征。作者还注意到不溶性p-tau蛋白和缺氧小鼠nrf2水平升高(Rojo et al., 2017)。本研究提示Nrf2缺乏与衰老/AD密切相关。这些来自不同实验室的发现支持了Nrf2可能在脑衰老和AD之间起分子连接作用的假设(Branca et al., 2017)。大量证据表明Nrf2功能障碍在认知障碍和AD的发病机制中起作用。Branca等人报道,在AD的APP/PS1小鼠模型中,Nrf2的缺失显著加重了包括空间学习和记忆在内的认知缺陷(Branca et al., 2017)。Nrf2还可以通过上调参与大自噬(Pajares et al., 2016)和伴侣蛋白介导的自噬(Pajares et al., 2018)的基因表达,帮助清除淀粉样蛋白前体蛋白(APP)和tau蛋白。这些发现可能解释了在app损伤和tau损伤的神经元中Nrf2水平的升高,并提示Nrf2激活可能在疾病进展的轻度认知障碍阶段很快发生。最近,Uruno等人证明Nrf2的诱导通过抑制OS和神经炎症改善了AD小鼠模型中的认知障碍(Uruno et al., 2020)。几种Nrf2激活因子在终止AD关键发病机制中也显示出强大的活性,包括抗抑郁药物和p-tau,以及对神经炎症和线粒体功能的调节(Ikram et al., 2019; Yang et al., 2018)。这些结果提示Nrf2是AD重要的治疗靶点。除了已知的Nrf2在维持细胞氧化还原稳态中的作用外,我们还进一步了解了Nrf2与AD关键发病因子的相互作用。
4.1. Nrf2和Aβ
AD的组织学特征是细胞外Aβ斑块沉积(Irvine et al., 2008;Selkoe and Hardy, 2016)。Aβ肽类(~4 kDa)是通过内含体中BACE1和γ-分泌酶顺序裂解APP产生的。BACE1是产生Aβ的必要物质,少量的BACE1的增加会引起一个显著的产量的增加。与分泌酶不同的是,BACE1的水平在AD中被发现升高,这表明它在AD中对细胞积累起重要作用。因此,BACE1的抑制剂正被寻求作为AD的治疗药物。Aβ易于聚集成寡聚体、原纤维和纤维形式的β-折叠构象。Aβ聚集的过程也涉及ROS的产生,导致神经元死亡(Andersen, 2004; Klein et al., 2001)。在AD大脑中发现了几种形式的Aβ,尤其是疏水性较强的Aβ,例如Aβ42,由于其高聚集倾向(Thal et al., 2015)。有证据表明,不同池中的Aβ数量和溶解度可能与疾病状态密切相关(Murphy and LeVine, 2010)。一些靶向聚合体或抑制BACE1的药物正在临床试验中,但它们都没有获得美国食品和药物管理局(FDA)的批准(Dong et al., 2019; Yan and Vassar, 2014)。
APP/PS1小鼠Nrf2基因删除显示炎症反应增强和细胞内APP、β42和β40水平升高。Nrf2缺失的APP/PS1小鼠神经元也显示了核内体、溶酶体和多泡小体的增加(Joshi et al., 2015)。这些发现为Nrf2在APP/ Aβ处理和积累以及自噬功能障碍中的作用提供了线索。Kerr等人首次提供了体内证据,证明通过特异性抑制Keap1激活Nrf2,可以防止AD诱导的Aβ42神经元毒性(Kerr et al., 2017)。Yu等人的一项研究也显示,当他们在AD动物模型中使用Nrf2激活剂isoastilbin时,也有类似的效果,并且他们发现Aβ42和 p tau表达降低(图4) (Yu et al., 2019)。Li等证明西格列汀和槲皮素的结合能够降低大鼠的Aβ表达水平(Li et al., 2019b)。Bahn等发现Nrf2通过与BACE1启动子中的AREs结合抑制了AD动物模型中BACE1的表达。Nrf2的激活减少了BACE1转录和Aβ产物的生成,并改善了认知缺陷。另一方面,Nrf2的缺失增加了BACE1的表达和Aβ生成,并加重了认知障碍。作者还证明,Nrf2对BACE1的调控与ROS抑制无关(Bahn et al., 2019)。本研究不仅将Nrf2的功能与Aβ生成联系起来,还揭示了Nrf2抑制BACE1的能力。有趣的是,该研究还发现Nrf2并不影响细胞分泌酶的活性或表达。这些发现表明,Nrf2的激活不仅抑制了Aβ的产生,还减少了其聚集所造成的损伤。
4.2. Nrf2和p-tau
MAP家族特别是MAP2和tau调节神经元中不同的细胞骨架过程(Amos and Schlieper, 2005)。Tau蛋白是一种55 kDa的蛋白,具有85个以上的翻译后修饰位点,如磷酸化、乙酰化和泛素化。虽然其正常功能需要翻译后磷酸化,但过度磷酸化会破坏其活性(Li and Gotz, 2017; Marciani, 2018; Morishima-Kawashima and Ihara, 2002)。Tau病理的发生是由于tau蛋白的错误折叠导致了β-折叠纤维的产生和积累。Tau病理和β-折叠纤维积累是包括AD在内的几种神经疾病的主要发病机制(Almansoub et al., 2019)。Tau病理被认为是AD认知能力下降和神经退行性变的主要驱动因素(Aschenbrenner et al., 2018; Hanseeuw et al., 2019)。
Jo等研究表明Nrf2可通过诱导核点蛋白52 (nuclear dot protein 52, NDP52)降低p-tau水平。NDP52是一种自噬接头蛋白,其诱导可促进自噬介导的p-tau蛋白降解(图4)。Jo等人也揭示了Nrf2活性的机制,即Nrf2与NDP52启动子中的AREs结合(Jo et al., 2014)。最近,Tang等报道Nrf2调节选择性自噬过程,可促进tau的清除。他们观察到,与年轻的Nrf2−/−动物相比,衰老Nrf2−/−动物中BAG3、NBR1、NDP52和p62的表达显著减少,这表明Nrf2在衰老过程中对维持这些基因的适当表达水平起着关键作用(Tang et al., 2018)。许多Nrf2激活剂,包括萝卜硫素(Kim et al., 2013)、苯福硫胺(Tapias et al., 2018)、富马酸二甲酯(Cuadrado et al., 2018)、大蒜素(Zhu et al., 2015)和mini-GAGR (Murphy et al., 2018)对p-tau AD动物模型显示出积极作用。
4.3. Nrf2 和神经炎症
小胶质细胞是常驻的免疫巨噬细胞样细胞,它们在急性感染和/或毒性损伤时的激活可能有助于清除受损细胞。在AD中,小胶质细胞可能被长期激活,导致有害的促炎细胞因子和ROS/RNS的持续分泌。这些过程导致神经退行性变。神经胶质细胞,位于Aβ斑块区域,表明激活的NF-κB、以及促炎细胞因子和趋化因子水平增加( Yin et al., 2017)。同时, NF-?B被发现调控BACE1表达 (Chen et al., 2012)。Terada等观察到tau病理与AD患者海马旁神经炎症呈正相关(Terada et al., 2019)。提示神经炎症可能在AD的发生发展中起重要作用。因此,神经炎症的消除可以减轻AD的进展和症状(Yang et al., 2019)。Nrf2和NF -κB之间的关系呈现出“阴阳”的概念。尽管在操作系统被激活,Nrf2诱导基因主要是抗炎(包括CD36、IL-17D)而NF –κB作用相反(增加IL-6、COX2和iNOS)。在这种背景下,Nrf2和NF-κB之间的不平衡对AD的发病机制可能是重要的。因此,Nrf2可能在AD中发挥重要的抗神经炎症作用。几项研究证实,Nrf2的激活可以抑制因注射Aβ 或tau引起的小胶质细胞激活,并进一步保护小胶质细胞引发的神经元丢失(Seo et al., 2017; Tom et al., 2019)。
4.4. Nrf2和线粒体功能障碍
线粒体是一种灵活的超微结构细胞器,它控制着关键分子元素的生物能量。线粒体的整体动态本质是由融合和分裂活动控制的。通过抑制线粒体破碎,融合可以延迟细胞凋亡的发生,而分裂对细胞凋亡有积极作用。线粒体质量控制过程中的失衡可能导致线粒体功能障碍(Cheng and Bai, 2018)。在Nrf2敲除小鼠中观察到年龄相关的线粒体OS以及线粒体功能受损,而Nrf2的激活增强了线粒体功能(Kitaoka et al., 2019)。Nrf2激活可增加蛋白酶体活性,而蛋白酶体活性可增强动力蛋白相关蛋白1 (Drp1)的降解(Sabouny et al., 2017)。Drp1是dynamin超家族的一种大型GTPase,它是促凋亡蛋白Bax线粒体转位所必需的酶,并与之结合以增强线粒体外膜通透性。这一过程促进细胞色素c的释放(Wang et al., 2015)。限制线粒体分裂蛋白的水平有利于线粒体低灌注,从而维持线粒体质量控制系统的功能。 Nrf2还控制线粒体抗凋亡蛋白(如Bcl-2和Bcl-xL)的表达、线粒体动力学、包括生物发生,通过与过氧化物酶体增殖激活受体-r 共激活剂 1a (PGC-1a)相互作用,以及通过p62依赖、PINK1/ parkino不依赖机制实现线粒体自噬(East et al., 2014; Merry and Ristow, 2016; Navarro et al., 2017)。AD早期出现线粒体功能障碍,导致神经退行性变,促进突触损伤和细胞凋亡。 、APP、Aβ和p-tau可干扰线粒体成分,导致线粒体动力学异常、生物发生缺陷、自噬减少、线粒体自噬减少、ATP生成受损和氧化损伤增加(Reddy et al., 2018; Sotolongo et al., 2020)。Nrf2可能减少这些损伤并维持线粒体功能(Fu et al., 2018)。Nrf2介导的Drp1降低也可能降低p-tau水平,提高突触活性(Cieri et al., 2018; Kandimalla et al., 2016)。几种Nrf2激活因子能够改善不同AD模型中的线粒体功能障碍(Li et al., 2019a; Liu et al., 2019; Tapias et al., 2018)。Nrf2在线粒体保护中的作用是Nrf2激活在AD治疗中的额外有益作用。
4.5. Nrf2和乙酰胆碱酯酶
乙酰胆碱(ACh)是一种有机神经递质,在神经末梢释放。乙酰胆碱乙酰转移酶(ChAT)以乙酰辅酶A和胆碱为底物在胆碱能神经元中产生乙酰胆碱。乙酰胆碱释放后,通过乙酰胆碱酯酶代谢成胆碱和醋酸盐。在AD患者的大脑中,已经观察到ACh水平的缺乏和AChE水平的增加。乙酰胆碱酯酶通过快速水解乙酰胆碱能突触终止冲动传递。因此,最小化活性乙酰胆碱酯酶水平可以增强胆碱能功能(Osama et al., 2017; Tabet, 2006)。尽管目前尚无明确证据表明Nrf2对乙酰胆碱酯酶(AChE)的影响,但在AD小鼠模型中,几种Nrf2激活因子均对胆碱能功能产生了积极影响,并降低了乙酰胆碱酯酶(AChE)水平,显著提高了乙酰胆碱酯酶(ACh)和乙酰胆碱酯酶(ChAT)水平(Huanga et al., 2019; Li et al., 2019c; Yu et al., 2019)。假设这些作用可能,至少部分是由于Nrf2的抗凋亡作用,可以防止神经元的丢失。Zhang等人发现,在细胞凋亡开始时,在细胞质中发现AChE,然后在细胞核或凋亡小体中发现,并细胞死亡。此外,凋亡细胞中表达的AChE与突触型AChE相同。此外,使用药理抑制剂或反义(阻断乙酰胆碱酯酶的表达)来最小化活性乙酰胆碱酯酶的水平,可以抑制细胞凋亡。他们的结论是,AChE可能是细胞凋亡的标志物和调节因子(Zhang et al., 2002)。这可能解释了AD大脑中大量细胞凋亡导致的高水平疼痛。Nrf2的抗凋亡作用可减少胆碱能神经元的丢失,从而维持乙酰胆碱的产生,使神经认知能力下降最小化。然而,对于AChE在细胞凋亡中的作用以及Nrf2的激活对AChE功能和分布的影响还需要进一步的研究。
5. AD中Nrf2相关通路的研究
一些研究也对AD中的Nrf2相关通路进行了研究,我们在此简要回顾其中一些。p62是一种细胞内信号蛋白,参与多种细胞环境。它还通过自噬参与聚合蛋白的降解。对p62基因敲除小鼠的研究表明,它们表现出一些类似AD的神经退行性变特征。郑等研究了AD模型中p62- Keap1-Nrf2关系,发现AD中p62水平降低可能与Nrf2水平降低有关。作者还推测这可能是AD中tau蛋白过度磷酸化和随后的神经元损伤的原因(Zheng et al., 2012)。Gu等人指出,Nrf2介导的p62信号通路的激活可通过激活自噬来改善由Aβ42损伤引起的细胞死亡(Gu et al., 2018)。Xu等人证实了p62在自噬介导的对致病性微管相关蛋白tau (MAPT)的清除中发挥的作用,并通过MAPT阻断了神经纤维缠结的积累和病理扩散。他们还表明,p62介导的对不溶性突变体MAPT的清除是非泛素依赖的(Xu et al., 2019)。
多项研究表明,AD中GSK-3β水平升高(Leroy et al., 2007; Ochalek et al., 2017; Pei et al., 1997; Virpi Talman et al., 2016)。GSK-3β抑制蛋白活性的增加直接参与了磷酸化导向的Nrf2的降解(图4)。GSK-3β抑制蛋白的基因过表达导致神经元丢失和记忆缺失,并参与了小鼠tau蛋白的过度磷酸化(Engel et al., 2006; Hernandez ´ et al., 2002)。Tideglusib是GSK-3β抑制因子,能够通过最小化tau蛋白磷酸化和减少淀粉样蛋白沉积来改善认知(Sereno et al., 2009)。Cuadrado等研究表明,富马酸二甲酯可以抑制Keap1和GSK-3β细胞,使Nrf2发生两层上调。在动物模型中,富马酸二甲酯能够显著降低tau蛋白磷酸化,并提供抗tau病理的保护(Cuadrado et al., 2018)。这一发现鼓励了对这类化合物的研究,它们可能对神经退行性疾病的治疗有用。
p38 MAPK在AD中被证实上调(Sheng et al., 2001; Sun, 2003)。除了降低Nrf2核易位外,它还可能通过OS激活Aβ(Giraldo et al., 2014)。最近的研究表明,p38的减少和Nrf2水平的升高可以消除SH-SY5Y细胞的神经毒性和OS (Amin et al., 2017a; Bach et al., 2011)。AD中NF-κB-p65的水平增加 (Terai et al ., 1996)。几个不同的AD模型研究表明,抑制NF-κB可以调控的表达Nrf2(Zhao et al., 2018)。Tom等人表明gedunin, Nrf2活化剂,可以通过抑制NF-κB及减少神经炎症,保护细胞免受Aβ42 寡聚体诱导的神经毒性 (Tom et al., 2019)。
6. Nrf2激活剂用于AD临床试验的影响
自1906年AD出现以来,只有五种药物被FDA批准用于临床治疗。这五种药是他克林,利瓦斯明,加兰他敏,多奈哌齐和美金刚胺。前四种药物是乙酰胆碱酯酶抑制剂,可以改善认知功能。美金刚胺是一种n -甲基- d -天冬氨酸(NMDA)受体拮抗剂,常用于治疗中重度AD。然而,这些药物都不能终止或逆转AD的潜在进展。它们只能暂时改善认知功能。他克林、利瓦斯明、加兰他明和多奈哌齐会产生一系列副作用,包括呕吐、腹泻、恶心、头晕和头痛。不幸的是,到目前为止,在AD的临床试验中,没有一种影响到Aβ和/或p-tau的分子显示出显著的作用(Dong et al., 2019)。所有这些因素使得新的治疗策略更具吸引力。如上所述,Nrf2可以通过直接和间接的方式干预Aβ和p-tau。此外,大多数AD患者是老年人,因此他们也可能患有与AD有共同病理特征的其他年龄相关疾病。Nrf2激活被发现对几种与年龄相关的疾病有用(Deshmukh et al., 2017; Francisqueti-Ferron et al., 2019; Silva-Palacios et al., 2018)。多靶点治疗被提出,因为乙酰胆碱酯酶抑制剂与抗氧化剂或NMDA受体拮抗剂一起给予治疗效果更好(Mangialasche et al., 2010)。但在这种情况下,使用单一的Nrf2激活剂可能对几种疾病有益,因此可能降低成本、副作用和药物相互作用。许多天然和合成的Nrf2激活剂已在神经退行性疾病的临床前研究或模型中得到验证(Dinkova-Kostova et al., 2017; Hou et al., 2019; Hou et al., 2018; Peng et al., 2019a; Peng et al., 2019b; Peng et al., 2015c; Yao et al., 2015)。最近,一些纳入AD临床试验的药物被证实能够激活Nrf2(表1)。2014年,苯磷硫胺被纳入AD临床试验(NCT02292238)。4年后,Tapias等人在体内模型中表明,苯磷硫胺可以激活Nrf2,并降低P301S AD中的p-tau (Tapias et al., 2018)。同样,DL-3-n-丁基苯酞(NCT02711683) (Qi et al., 2018)和培哚普利 (NCT02085265) (Kamel et al., 2020)的Nrf2激活作用最近也得到了验证。从十字花科植物中分离得到的萝卜硫素进入AD临床试验(NCT04213391)可能是开发基于Nrf2激活的AD新药的一个很好的步骤。萝卜硫素通过与Keap1的Cys151相互作用,也是Nrf2亲电激活剂。萝卜硫素具有神经保护作用,干预Aβ 和 p tau,并改善急性AD的认知缺陷(Kim et al., 2013)。目前,萝卜硫素有20多个临床试验。然而,萝卜硫素在室温下相对不稳定。因此,它的合成类似物SFX-01 (Evgen Pharma开发的药物)备受关注。这些能力使萝卜硫素及其类似物成为AD药物开发的重要药物。
在FDA批准的可激活Nrf2的药物中,富马酸二甲酯被认为是治疗AD的有前景的药物。2013年,富马酸二甲酯被FDA批准用于治疗多发性硬化症。富马酸二甲酯在不同的AD模型中均表现出积极的作用,因此被认为是药物重新定位的最佳选择之一。总的来说,半不饱和羰基化合物是研究最多的可激活Nrf2的化合物。这类化合物被设计通过与Keap1的半胱氨酸残基结合来干扰Keap1- nrf2的相互作用(Peng et al., 2015b)。几种自然产生的药剂,包括xanthohumol, 6 dehydrogingerdione, cardamonin, 6-shogaol and chlorogenic acid,属于这一家族,并已被我们实验室鉴定为通过激活Nrf2来发挥神经保护能力(Bai et al., 2019; Peng et al., 2017; Peng et al., 2015a; Yao et al., 2014; Yao et al., 2019)。
目前,有数十种天然和合成的Nrf2激活剂可用于进一步深入研究其在AD模型中的作用。表2总结了参与不同临床试验并证明对AD动物模型有积极影响的化合物。这些化合物可能是药物开发的良好起点。化学修饰也可能有助于改善这些分子的化学和药理特性,从而达到更好的治疗效果。
7. Nrf2治疗AD药物开发的挑战
找到一种有效且相对安全的治疗AD的新药不是一件容易的事。此外,通过靶向Nrf2探索这种新的治疗方法也使这项任务更具挑战性。在这里,我们讨论几个问题和考虑开发Nrf2激活剂的临床应用。
7.1. 选择性
Nrf2抑制蛋白Keap1是一个富含Cys的蛋白,有27个Cys残基,其中一些高度反应。Keap1的这一特性促使许多研究小组设计能够与Keap1的Cys残基硫醇基团形成共价键的共价抑制剂。在27个Cys残基中,Cys151、Cys273和Cys288得到了高度关注。Cys273和Cys288在基础和应激条件下Nrf2的调控中都发挥了重要作用。另一方面,Cys151在应对各种应激条件方面具有重要作用(Suzuki et al., 2013)。许多亲电物种能够与Keap1的Cys残基形成共价相互作用,从而抑制Nrf2泛素化。用亲电修饰剂随机或选择性地攻击Keap1 Cys残基被称为“半胱氨酸编码”(Cuadrado et al., 2019; Fao et al., 2019)。不幸的是,攻击Keap1中的Cys残基并不意味着不能攻击其他富含半胱氨酸的蛋白质。这种缺乏选择性是开发共价Keap1修饰剂的主要问题(Suzuki et al., 2013)。人们正在努力发现更有选择性的化合物,蛋白质-蛋白质相互作用(PPI)抑制剂也在选择之列。CPUY192018是通过阻断Nrf2和Keap1的PPI来激活Nrf2的最成功的例子之一(Lu et al., 2019)。基于更大的支架结构可能提供更好的选择性的假设,也研究了五环迈克尔受体的氰烯酮三萜。Bardoxolone甲基就是这类化合物的一部分,目前正在进行四个临床试验(Cuadrado et al., 2019)。Nrf2可以通过其他不同的策略激活,从而提高选择性。Nrf2激活的途径之一是使用竞争性抑制剂攻击Keap1-Nrf2的结合位点。这一策略是实用的和高度选择性的,因此它可能是减少副作用的一个很好的替代方法(Madden and Itzhaki, 2020)。另一种方法是靶向BTB结构域中的Keap1二聚作用(Zhuang et al., 2009)。BTB结构域也参与了Keap1和Cul3的结合,针对这种Keap1-Cul3的相互作用也可能导致Nrf2的激活。Keap1 IVR域中发现的靶向3-box也可能干扰Keap1和Cul3的结合(Cleasby et al., 2014)。用新的激活剂靶向这些不同的位点可以增加选择性,从而减少潜在药物的副作用。此外,靶向激活AD脑损伤区域的Nrf2是另一个挑战,进一步修饰这些化合物可能是提高选择性的一个有希望的策略。
7.2. AD实验模型
在药物发现领域,实验模型是非常重要的。这些模型在阐明疾病的病理生理原因和测试候选药物的临床前后果方面至关重要。在AD的案例中,对多个物种的神经病理学调查显示,AD似乎是人类特有的(Drummond and Wisniewski, 2016)。对包括恒河猴、狒狒、黑猩猩、食蟹猕猴和狐猴在内的非人类模型的研究表明,它们具有AD的一些共同特征,有助于解开AD病理的一些模糊之处。不幸的是,在这些模型上进行研究可能会花费很长时间,毕竟,它们可能会随着它们表现出的特征而变化,因为它们没有受到人类AD的折磨。AD治疗学的临床试验失败率极高(>99%),这可能与缺乏合适的AD动物模型有关。在人工诱导非人类AD模型方面已经做了很多工作,包括注射模型和转基因小鼠模型。这些模型可用于数百种潜在药物的AD临床前测试(Esquerda-Canals et al., 2017)。不幸的是,在数百种临床前成功的候选药物中,只有5种获得了市场批准。在临床试验中未能取得成功可能是因为这些模型考虑了毒性APP和/或tau突变蛋白的表达,而忽略了其他与疾病相关的下降,如与Nrf2相关的稳态功能。虽然Nrf2敲除小鼠表现出与衰老和AD共同的特征(Rojo et al., 2017),但该模型是否与AD病理更相关还需要更多的努力来验证。
7.3. 能否通过血脑屏障(BBB)
为了达到预期的药理学效果,需要考虑许多候选药物的因素,包括生物利用度、代谢稳定性、溶解性和细胞通透性。此外,药物应表现出更多的亲和力,或至少可以达到目标器官的足够剂量(Krogsgaard-Larsen et al., 2005)。因此,要激活AD神经元中的Nrf2,药物必须以足够的浓度到达AD脑损伤区,使其能够发挥作用。血脑屏障是保护中枢神经系统的天然屏障,通过阻止神经毒性血浆物种、血细胞和病原体进入大脑。不能跨越血脑屏障的药物获得AD临床试验成功的机会很低(Pardridge, 2019)。细胞培养研究可能大大高估了测试药物的活性。由于生物利用度低,这些候选药物在临床试验中可能表现出令人失望的结果。分子量< 450道尔顿、极性表面积< 90 A2和氢键供体数< 3的药物更有可能穿透血BBB (Hanson and Frey, 2008)。这些通用指南为中枢神经系统药物的设计和开发提供了初步考虑。然而,血脑屏障的侵彻能力必须通过实验来评估。因此,需要在亲脂性、分子构型和分子大小之间实现神奇的结合和平衡,才能使药物通过血脑屏障。
7.4. 确定给药方案
关于Nrf2激活治疗AD的另一个考虑是,老年人和不太健康的受试者对Nrf2激活剂的反应降低(Hammer et al., 2018)。因此,给药剂量必须考虑患者的年龄和健康状况。由于Nrf2与其靶基因相比半衰期较短,因此也应通过分析药物分布和Nrf2基因在可接近细胞或组织中的表达特征来间接推断合适的给药方案(Cuadrado et al., 2019)。
8. 结论
Nrf2是抗氧化系统的主要控制器,负责II期相解毒酶和抗氧化蛋白的组成性表达。大量的生物分子参与了Nrf2的调控,这说明了这一途径的复杂性。AD中Nrf2、p62 GSK-3β,p38和NF -κB-p65水平发生改变。大量证据表明,Nrf2具有在各种AD模型中降低OS、炎症和线粒体功能障碍的能力(图5)。有趣的是,Nrf2也可以抑制BACE1的表达,并改善Aβ介导的毒性。Nrf2还能降低AD模型中p-tau和认知缺陷的水平。几种Nrf2激活剂在不同的AD动物模型中显示了良好的效果。基于这些研究,以及并没有FDA批准新的药物用于临床治疗AD, Nrf2激活似乎是一种新颖而有吸引力的治疗方法。然而,我们并不认为这种治疗方法会比其他方法更容易。Nrf2通路本身复杂,可通过不同的机制激活。此外,Nrf2激活剂的开发也面临着许多挑战。用亲电共价抑制剂攻击Keap1中的Cys残基是Nrf2激活的最常见机制。不幸的是,这种类型的活化剂通常缺乏选择性。低选择性药物有望引起广泛的副作用。有几种方法可以用来解决这个问题,包括开发PPI抑制剂和非共价Keap1抑制剂。缺乏理想的AD动物模型是AD药物发现领域的另一个障碍。到目前为止,关于Nrf2激活在人类AD大脑中的作用的信息很少。最近在AD临床试验中加入的Nrf2激活因子,如萝卜硫素,可能为Nrf2激活在AD中的结果提供有价值的信息。
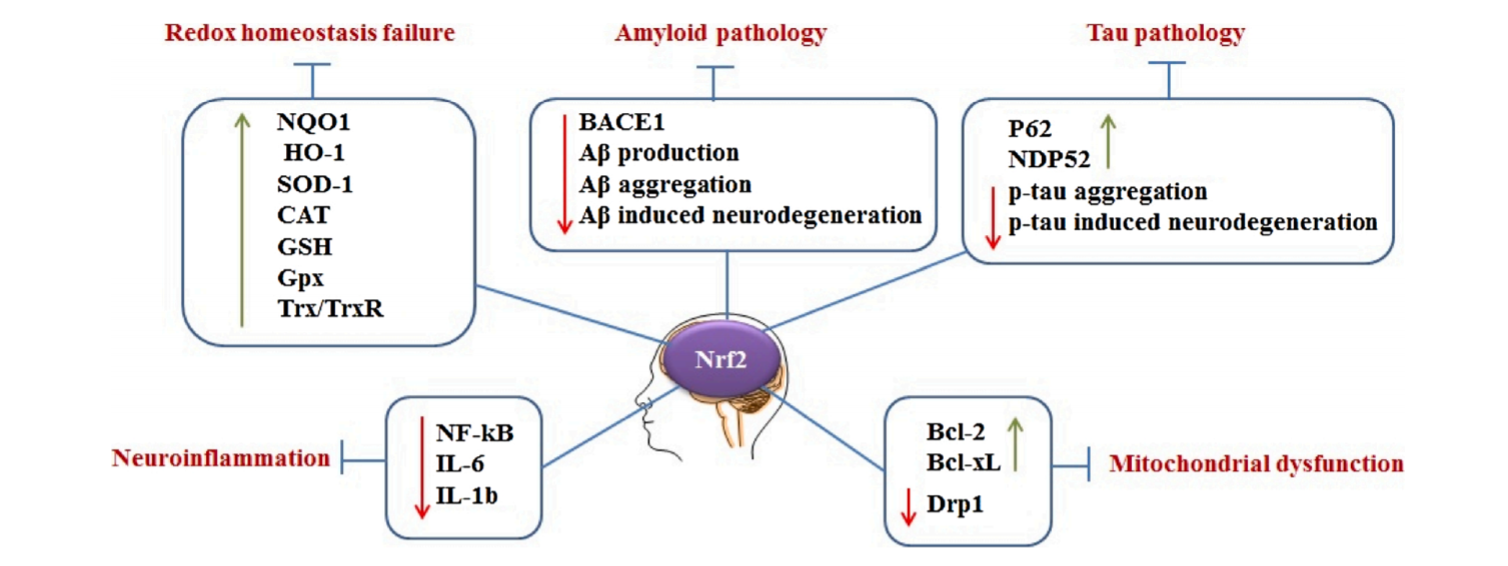
图1:AD大脑遭受氧化应激。(a)线粒体功能障碍可能导致ROS产生增加和细胞凋亡。(b)在AD中观察到广泛的脂质过氧化。(c) AD大脑还显示出神经毒性微量元素如铁、铝、铜和汞的水平提高,这些元素可诱导自由基的形成(铁就是一个例子)。(d)高水平Aβ(Aβ42;PDB: 5kk3)也可促进AD脑内的氧化应激。(e)高水平的ROS可能攻击DNA,因此AD脑显示更多的氧化DNA(鸟嘌呤为例)。
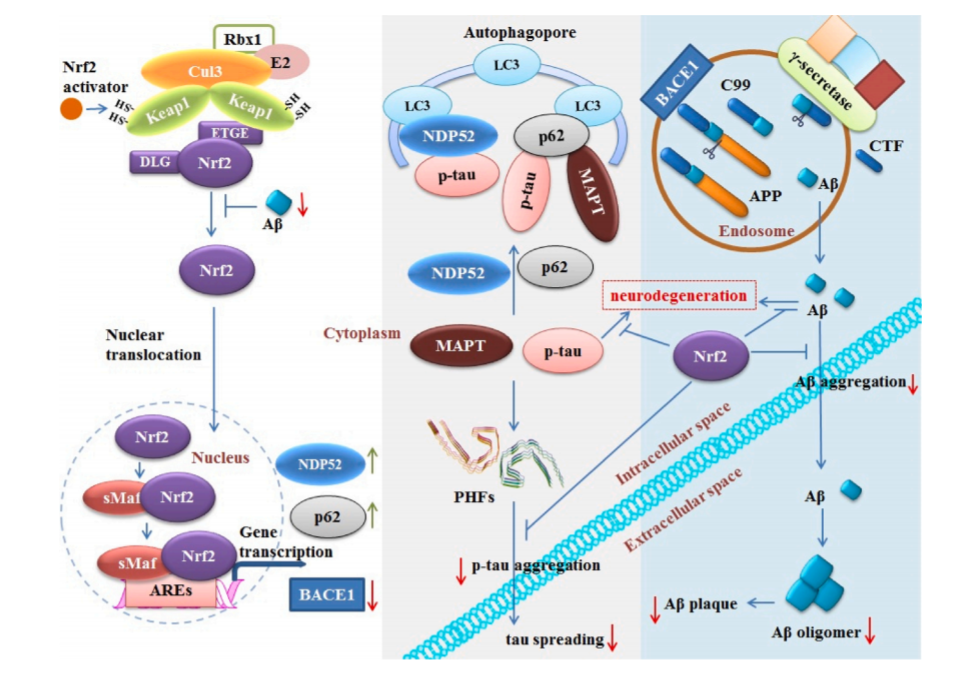
图2:Nrf2和Keap1蛋白不同结构域的示意图。(a) Nrf2的七个Neh域及其功能。(b) Keap1的五个代表性区域和几个重要的半胱氨酸传感器在不同区域发现。
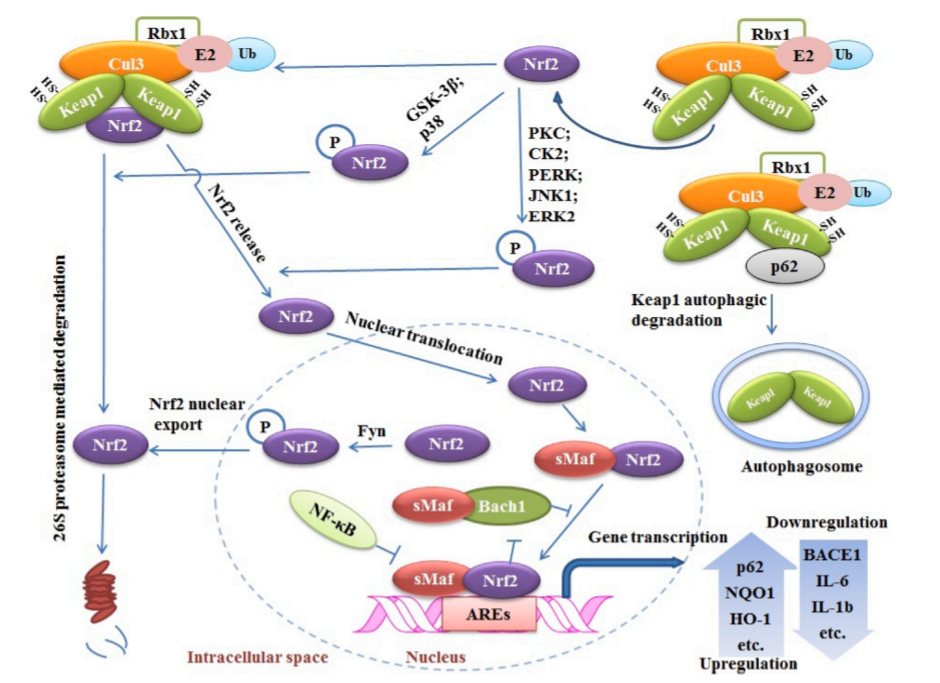
图3:Nrf2的keap1依赖性和非依赖调控机制。在基础条件下,Keap1-Cul3复合物与Nrf2结合并介导其泛素化和降解。p62可能通过与Keap1结合,导致Keap1的自噬降解,从而减少了这一过程,从而增加了细胞核中Nrf2的积累。GSK-3β和p38 MAPK可使Nrf2在Neh6区域磷酸化并有利于其降解。相反,PKC、CK2 PERK、JNK1和ERK2介导的Nrf2磷酸化促进了Nrf2与Keap1的分离和核积累。在细胞核中,Nrf2与转录共激活因子Mafs形成异源二聚体,促进与ARE的稳定结合并增强基因转录。Fyn激酶可能使Nrf2在Tyr568处磷酸化,有利于其核出口。Bach1与Nrf2竞争结合导致Nrf2转录能力下降。NF-κB也通过与ARE结合与Nrf2竞争, 下调其转录活性。
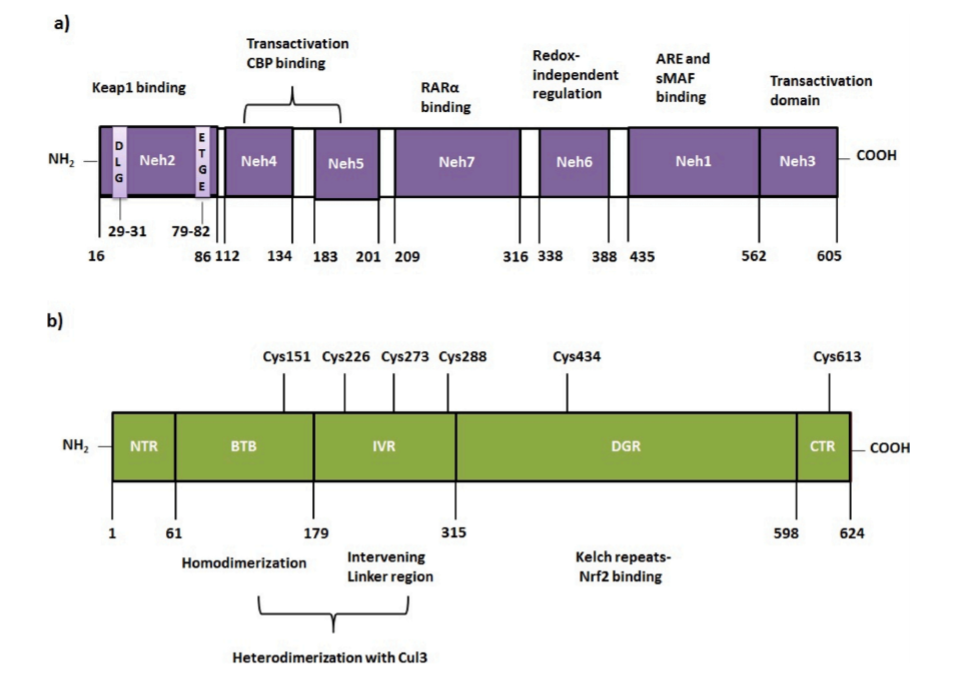
图4:AD中Nrf2与一个高亮区域(蓝色)和p-tau(灰色)相互作用的示意图。Aβ通过内含体中BACE1和γ-分泌酶的蛋白水解功能从APP中衍生出来。Aβ分泌入组织间液,可导致Nrf2水平降低。Aβ可聚集成寡聚体和原纤维,对细胞功能有许多影响,包括突触活性受损和脑毛细血管血流量受损。Nrf2的激活降低了BACE1的转录水平,从而降低了Aβ的产生。Nrf2还可能降低聚合Aβ的水平,从而减少AD淀粉样蛋白的病理。错误折叠的MAPT和p-tau蛋白的积累可能导致成对螺旋丝的形成(PHF;PDB: 6HRE),随后跨突触传播到远处的神经元。Nrf2的激活可能会增加参与选择性自噬过程的p62和NDP52的水平。P62还可以促进不溶性MAPT的降解,从而降低神经纤维缠结(NFTs)水平。Nrf2可能预防p-tau诱导的神经退行性变,也可能减少p-tau的聚集。因此,Nrf2的激活导致AD tau的病理减少。
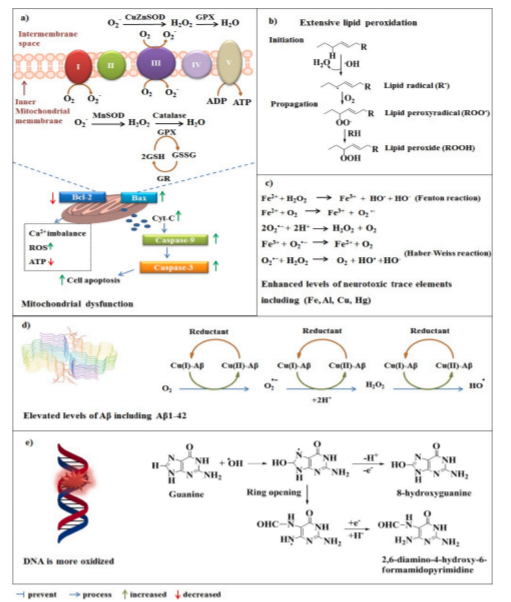
图5:Nrf2在AD中的作用的简化说明。Nrf2通过(上调/下调)调控多个下游靶点来抑制AD的几个关键致病特征。
翻译:
Nrf2:阿尔茨海默氏病治疗中的黑马
摘要:阿尔茨海默氏病(AD)是一种年龄依赖性神经退行性疾病,是痴呆症的主要原因。AD的常见特征包括淀粉样β肽(Aβ)聚集,高水平的高磷酸化tau蛋白(p-tau)和氧化还原稳态失败。迄今为止,所有针对Aβ和/或p-tau的药物临床试验均失败了。在AD大脑中已经观察到转录因子Nrf2(核因子-类胡萝卜素2-p45衍生因子2)及其驱动基因(NQO1,HO-1和GCLC)的表达下降,以及Nrf2相关途径的改变。Nrf2在维持细胞氧化还原稳态和调节炎症反应中起关键作用。Nrf2激活还提供针对日益增加的病理(包括神经退行性疾病)的细胞保护作用。这些证据表明,Nrf2激活可能是一种新颖的AD治疗选择。有趣的是,最近的研究还表明,Nrf2会干扰AD中的几种关键致病过程,包括Aβ和p-tau途径。本综述旨在深入了解Nrf2在AD中的作用。此外,我们还讨论了有关Nrf2激活剂用于AD治疗的进展和挑战。
关键词:阿尔茨海默病、Nrf2、 Keap1、β淀粉样蛋白肽、Tau蛋白、氧化应激
1. 前言
神经元主要参与学习、思考、记忆和计划(Carter等人,2019)。神经退行性疾病的主要原因是多种病理因素导致神经元功能和生存能力的逐步下降。这些多因素神经退行性疾病正在影响全球数百万人(Calderon-Garciduenas和Duyckaerts,2017)。神经退行性疾病,例如阿尔茨海默氏病(AD),被证明与蛋白质折叠错误、线粒体损伤和氧化应激(OS)有关。AD是一种不可逆的进行性脑部疾病,会逐渐发生并导致记忆力减退以及思维和行为下降(Lu等人,2018年; Ossenkoppele等人,2015年)。AD被认为是痴呆症的主要原因,这是至少两个认知领域的逐步衰退,并导致正常的社交或职业活动残疾。AD通常会影响老年人,但它不是正常的衰老缺陷。AD的特征包括淀粉样β肽(Aβ)的聚集、高磷酸化tau蛋白(p-tau)的积累、炎性介质的产生、OS以及胆碱能、突触和认知功能的障碍。遗传、环境因素和一般生活方式是与AD相关的病因因素的一部分(Long和Holtzman,2019)。当前,AD影响着超过5000万人,估计2050年全球发病率将达到1.5亿(Jannat等人,2019)。由于仅部分了解AD的发病机理,因此AD的治疗仍然是一个挑战(Park等人,2018)。许多III期临床试验都失败了,尤其是针对Aβ产生和聚集的试验。由于缺乏明显的疗效,最近终止了Crenezumab(NCT03491150)、lanabecestat(NCT02972658)和solanezumab(NCT01900665)临床试验。在寻找治疗AD的有效药物方面的系统性失败,促使科学家们探索新的策略来对抗这种疾病。
AD具有多种病理特征,例如大量的DNA氧化、严重的线粒体损伤、广泛的脂质过氧化作用、高水平的神经毒性微量元素以及Aβ的升高(Deibel等,1996; Huang等,1999; Wang等(2005年)。所有这些因素都会增加自由基或活性氧(ROS)的形成,因此,因此AD患者的大脑也受到OS的影响(图1)。在生理条件下,ROS的动态平衡受到ROS生成系统和细胞抗氧化网络的良好调控(Zhang等,2020; Zhang等,2019)。抗氧化防御系统包括几种酶和小分子,例如血红素加氧酶-1(HO-1)、超氧化物歧化酶(SOD)酶、谷胱甘肽(GSH)、过氧化氢酶(CAT)、硫氧还蛋白(Trx)、硫氧还蛋白还原酶(TrxR)、谷胱甘肽过氧化物酶(Gpx)家族和NAD(P)H:醌氧化还原酶1(NQO1)。在人类AD研究中,患者的大脑显示出核因子红系2相关因子2(Nrf2)、SOD1、CAT和Gpx的水平降低(Chenere等,2007; Mota等,2015)。由于Nrf2和这些重要的抗氧化酶含量低,AD大脑容易受到ROS的侵害。在这种情况下,ROS对大脑造成进行性和不可逆的损害。用抗氧化剂(例如维生素E、维生素C和雌激素)直接治疗可能在AD中发挥积极作用,但这些抗氧化剂在临床患者中的疗效仍处于初级阶段(Feng和Wang,2012)。通过Nrf2活化来增加抗氧化蛋白的水平已被认为是实现神经保护的更好方法(Buendia等人,2016; Chen和Maltagliati,2018)。 Nrf2诱导大约250个基因编码不同的细胞保护和解毒蛋白的表达。高水平的Nrf2可以缓解AD中ROS和/或线粒体功能障碍引起的损害(Eftekharzadeh等人,2010; Fu等人,2018)。最近的研究表明Nrf2可以预防AD中某些关键的早期致病过程,包括Aβ(Cuadrado等,2018; Han等,2019)。已知Nrf2可通过调节抗氧化蛋白和解毒蛋白发挥神经保护作用,但是Nrf2具有干扰β-分泌酶(BACE1)、Aβ和p-tau的能力是一个有趣的发现。该领域的一些优秀的研究是由不同的研究小组进行的(Bahn等人,2019; Kitaoka等人,2019; Sotolongo等人,2020; Tang等人,2018),在此,我们旨在回顾和讨论这些发现,以提高我们对Nrf2在AD中的作用的认识。理解Nrf2激活剂针对Aβ和p-tau的神经保护作用的潜在机制可能有助于新AD药物的设计和开发。此外,我们讨论了Nrf2激活剂作为AD潜在疗法的进展以及开发Nrf2激活剂的主要挑战。在药物设计过程中考虑这些问题可能会增加临床试验的成功率。
2. Nrf2的结构和功能
Nrf2蛋白是NFE2L2基因的产物,是研究最多的Cap 'n 'collar基区亮氨酸拉链(b-Zip)转录因子家族成员之一。Nrf2自1994年被发现以来,关于该转录因子的研究不断增多。学术界和产业界之所以对Nrf2如此感兴趣,很大程度上是因为Nrf2具有广泛的功能。Nrf2是细胞保护系统的主要调控器,即II相解毒酶和抗氧化剂 (Cuadrado et al., 2019)。Nrf2参与NADPH、NQO1、HO-1、胆绿素还原酶、谷胱甘肽家族和硫氧还蛋白系统的转录调控。基于Nrf2的转录程序的细胞保护作用使细胞适应并在应激条件下生存(Hou et al., 2018)。这些功能会改变代谢和生物能,从而影响中间代谢和线粒体功能(Kitaoka et al., 2019; Merry and Ristow, 2016)。由于Nrf2调控多种抗氧化酶的表达,其抗氧化作用已被广泛研究(Peng et al., 2015b)。Nrf2在自噬调控中的作用已经被许多实验室解决,他们观察到Nrf2激活导致自噬增加,而Nrf2缺乏导致自噬减少(Frias et al., 2020; Jo et al., 2014; Komatsu et al., 2010; Pajares et al., 2018)。激活自噬可增加溶酶体介导的蛋白降解和损伤细胞器及感染微生物的清除(Tang et al., 2018)。Nrf2还参与调控另一种类型的细胞死亡,即铁依赖性脂质过氧化引起的铁死亡。Fenton反应(图1c)可能部分解释了铁死亡对铁的依赖,因为具有氧化还原活性的铁池可以催化脂质过氧化,形成损伤,最终导致细胞死亡。与其他类型的细胞死亡相比,铁死亡表现出独特的形态、生化和遗传特征(Dixon et al., 2012)。由于脂质过氧化是铁死亡的核心特征,脂质过氧化物清除剂能有效中和铁作用(Homma et al., 2019; Matsushita et al., 2015)。有趣的是,到目前为止,所有涉及铁蛋白作用的基因都受到Nrf2的转录调控,包括但不限于Gpx4、Trx、TrxR、HO-1、铁转运蛋白和铁螯合酶。因此,Nrf2可以作为这一过程的主要调控因子。人们对探索铁死亡在包括AD在内的神经退行性疾病中的作用越来越感兴趣(Cong et al., 2019; Do Van et al., 2016)。然而,还需要进一步的研究来验证铁蛋白增多与神经退行性疾病的联系。Nrf2激活的另一个显著特征是炎症抑制。Nrf2的抗炎能力比预期的更为复杂, 它涉及Nrf2靶基因编码蛋白的转录上调(如白三烯B4脱氢酶) 以及编码的主要促炎细胞因子(如IL-6和IL-1b)基因的下调 (Dick et al., 2001; Kobayashi et al., 2016)。OS和炎症是包括癌症、心血管和神经退行性疾病在内的大多数人类病理的主要因素(Kwak and Kensler, 2010; Liby et al., 2012)。特别是Nrf2在神经退行性疾病、糖尿病和肾脏疾病中的作用是目前研究的热点 (Das et al., 2013; Fao et al., 2019; Matzinger et al., 2018; Yamawaki et al., 2018)。
不幸的是,到目前为止,Nrf2结构还没有被完全阐明。人类Nrf2蛋白由605个氨基酸组成。由七个区域组成,命名为Neh1-Neh7 (Nrf2-ECH同源性)。Neh1、Neh3和Neh6位于c端。另一方面(n -端)包含Neh2、Neh4和Neh5结构域。Neh7是由Wang等人发现的,与Neh5相邻。Neh1负责DNA结合(图2a) (Wang et al., 2013)。Neh2通过两个基序DLG和ETGE与kelch样ech相关蛋白1 (Keap1)相互作用(Nioi et al. , 2005)。Neh3是一个反激活区域,可以提高抗氧化反应元件(ARE)依赖基因的转录(McMahon et al. , 2004)。Nrf2的keap1独立降解主要由Neh6区域介导。Neh7可能与视黄酸受体(RAR α)结合,这种相互作用导致Nrf2-ARE信号通路的负调控(Wang et al. , 2013)。
3. Nrf2调控的分子机制
Nrf2调控大量基因的表达。同时,它也是大量生物分子的靶点。这些生物分子以不同的机制调控Nrf2功能。了解Nrf2的调控机制对于开发靶向Nrf2的新分子以及了解Nrf2的治疗作用具有重要意义。Kwak等人描述了Nrf2通过结合其启动子区域激活其表达的能力,这支持了Nrf2自我调节(autoregulation)的观点(Kwak et al., 2002; Miao et al., 2005; Rushmore et al., 1990)。Nrf2的调控除了自身调控外,主要分为keap1依赖性和非keap1依赖性调控。这里我们总结了Nrf2调控的机制(图3)。
3.1. Keap1依赖性调控
自从Itoh等人发现去除Nrf2的Neh2可以增加Nrf2活性以来,Keap1在Nrf2调控中的作用就已经为人所知(Itoh et al., 1999)。人Keap1(基因ID: 9817)蛋白是由624个氨基酸残基组成的富含半胱氨酸的蛋白,表观分子量为69.7 kDa (Itoh et al., 1999; Zipper and Mulcahy, 2002)。Keap1有5个具有不同功能的结构域,即N端区域(NTR;1-60),C末端区域CTR(599-624)、BTB(61-179)(Broad complex, Tramtrack and Bric a Brac)/POZ(poxvirus and zinc finger)、双甘氨酸重复区DGR (315-598) (double glycine repeat or Kelch repeats)及插入区IVR(180-314)(intervening region)。DGR和CTR一起被称为Keap1-DC,它对Nrf2的Neh2结构域的结合至关重要。人Keap1含有27个半胱氨酸(Cys)残基,但只有Cys151、Cys226、Cys273、Cys288、Cys434和Cys613对Nrf2激活至关重要(图2b) (Bryan et al., 2013; Canning et al., 2013; Canning et al., 2015)。
Nrf2是Keap1最具特征的靶蛋白之一。在基础条件下,Keap1有利于Nrf2蛋白酶体降解,以防止ARE的组成性激活(Huang et al., 2000)。人们提出了不同的模型来理解这种相互作用和调节的机制。铰链和插销模型是理解Keap1-Nrf2相互作用的首选模型(图4)。在这个模型中,Nrf2 ETGE基序首先与Keap1结合。ETGE结构域采用圆转构象,深深插入Keap1口袋,埋藏面积为420 Å2 (Abiko et al., 2011; Wakabayashi et al., 2004)。然后Nrf2通过其DLG基序在Keap1的第二个独立结合位点结合。通过对ETGE和DLG的比较,可以看出两者都与Kelch域口袋的底部相互作用,但其构象和亲和力不同。热力学和动力学分析表明,与Keap1-DLG较弱的相互作用相反,Keap1-ETGE是焓驱动的,开关速率较慢,导致高亲和力的相互作用(Hong et al., 2005; McMahon et al., 2003; Tong et al., 2007)。在激活模式下,DLG基序与Keap1分离,导致Nrf2的释放和转位到细胞核(Bloom and Jaiswal, 2003)。
Nrf2的另一个keap1依赖性调控是通过p62。p62蛋白也被称为死骨片1 (SQSTM1)。p62蛋白可以结合多聚泛素化蛋白和自噬机制,靶向自噬的特定载体(Kim et al., 2008; Pankiv et al., 2007)。Komatsu等证明p62通过与Keap1的相互作用破坏Keap1-Nrf2系统(Komatsu et al., 2010)。p62通过其与Nrf2 ETGE相似的STGE基序与Keap1结合。p62高表达导致Nrf2水平升高,反之亦然(Copple et al., 2010)。另一项研究也表明,p62的积累导致Nrf2的一致激活(Inami et al., 2011)。为了理解p62-Keap1结合模型,人们做了很多有价值的工作,结果证明该模型与Keap1的CTR重叠(Komatsu et al., 2010;Lau et al., 2010)。基于编码p62蛋白的mRNA的存在,缺失了与Keap1结合所必需的Keap1相互作用区域,Kageyama等人证明,该变异增加了Keap1的数量,并增强了Nrf2的泛素化。本研究表明,p62 pre-mRNA的剪接负调控小鼠Keap1-Nrf2通路(Kageyama et al., 2018)。然而,关于人类的这种变异还需要更多的研究。BRCA2(PALB2)的伴侣和定位剂二肽基肽酶3(DPP3)也通过ETGE基序与Keap1相互作用,激活Nrf2(Hast et al., 2013)。
3.2. 非Keap1依赖性调控
Nrf2中非keap1依赖性调控也被几个研究小组研究过。在这方面,Nrf2的翻译后修饰,包括磷酸化、泛素化和乙酰化得到了很好的表征。Nrf2含有许多苏氨酸、丝氨酸和酪氨酸残基,是潜在的磷酸化位点。几种激酶可能磷酸化Nrf2并调节其活性(Rojo et al., 2012)。蛋白激酶C (PKC)磷酸化Nrf2的Ser40 (Neh2),从而破坏与Keap1的关联,进而促进Nrf2转位进入细胞核(Huang et al., 2002)。最近, Fao等人表明,Nrf2在Ser40被c-src依赖激酶磷酸化激活,在Tyr311通过PKCδ磷酸化 (Fao et al ., 2019)。酪蛋白激酶II (CK2)在Nrf2特异序列中也有大约13个潜在的磷酸化位点(Neh4和Neh5) (Pi et al., 2007)。这些位点的磷酸化与易位进入细胞核的Nrf2相连接。研究发现,在CK2抑制剂存在的情况下,Nrf2易位降低(Patrick L. Apopa et al., 2008)。类似地,蛋白激酶R (PKR)样内质网激酶(PERK)介导了Thr80的磷酸化并有利于Nrf2的激活(Cullinan and Diehl, 2004)。糖原合成酶激酶-3蛋白(GSK-3磷酸化)使Nrf2的Ser335和Ser338位点磷酸化并导致其降解(Rada et al., 2011; Rojo et al., 2008)。GSK-3蛋白还作用于酪氨酸激酶Fyn的上游,可以磷酸化Nrf2中的Tyr568,导致Nrf2的核积累(Jain and Jaiswal, 2006,2007)。几种丝裂原活化蛋白(MAP)激酶也显示了不同Nrf2丝氨酸和苏氨酸残基的磷酸化能力。虽然,c-Jun n末端激酶1 (JNK1)和细胞外调节蛋白激酶2 (ERK2)介导的磷酸化诱导了Nrf2的激活,但p38通过磷酸化Nrf2促进keap1和Nrf2之间的联系,从而阻止其核易位(Keum et al., 2006)。Hrd1 (E3泛素连接酶)又称滑膜泛素连接酶。Hrd1对内质网相关降解(ERAD)至关重要。Wu等证明Hrd1与Nrf2的Neh4-5结构域相互作用导致Nrf2泛素化并降解(Wu et al., 2014)。CBP/p300介导的Nrf2赖氨酸残基乙酰化(Neh3/Neh1)增加了Nrf2的转录功能。另一方面,sirt1介导的去乙酰化降低了Nrf2信号(Kawai et al., 2011)。
核因子-κB (NF-κB)蛋白是一组结构相关转录因子,参与了大量的细胞过程。NF -κB可以抑制相关基因的转录,而几个Nrf2催化剂有能力抑制NF-κB活动(Nair et al ., 2008)。大量证据还表明,Keap1通过与IκB激酶-β(IKKβ)结合而在NF-κB通路的负调控中起作用,并赋予NF-κB泛素化和降解作用(Kim et al., 2010; Lee et al., 2009)。
Bach1是b-Zip转录因子的一部分,与小Maf蛋白形成异源二聚体,并作为Nrf2的诱导抑制因子。Bach1/Maf和Nrf2/Maf竞争性地共享同一结合位点(ARE的顺式元素)。因此,靶基因的抑制或激活可能发生。然而,Bach1的亚细胞定位依赖于Nrf2核输出信号和Crm1, Crm1由氧化还原敏感的细胞质定位信号(CLS)驱动,因此Bach1主要由Nrf2调控(Silva-Palacios et al., 2018)。Poganik等人引入了一种新的直接调控Nrf2 mRNA的转录后模型。他们报告说,HuR和AUF1 ,两个mRNA结合蛋白质,可以通过靶向mRNA 3?utr专门调控Nrf2。他们的结果表明,HuR可以促进Nrf2 mRNA的成熟并促进其核输出,而AUF1则稳定Nrf2 mRNA。他们还发现,Nrf2活性的转录后调控在整个脊椎动物水平上是保守的(Poganik et al., 2019)。这一发现为发现更多遵循类似调控机制的Nrf2调控因子奠定了基础。
几项研究已经鉴定出一些通过将Nrf2的mRNA靶向3´UTR来调节Nrf2表达的MicroRNA。这些Nrf2负调节剂的例子包括但不限于miR28,miR-153,miR-142-5p,miR-27a和miR-144(Narasimhan et al., 2012; Yang et al., 2011)。然而,Nrf2与其靶向miRNA之间的关系是高度复杂的,需要更多的研究来发现其他参与Nrf2调控的miRNA及其调控机制以及这种调控在AD发病机制中的作用。综合来看,参与Nrf2调控的多个因素可以为理解Nrf2参与不同疾病和紊乱的治疗提供线索(Cuadrado et al., 2019)。
4. Nrf2在AD中的作用
Nrf2在机体的不同器官(包括大脑)中表达,表达水平各不相同。代谢器官,特别是那些暴露于外部环境的亲电性和氧化剂的器官(如肺和肠) Nrf2水平较高(Riedl et al., 2009)。对人和小鼠大脑中枢神经系统(CNS)的研究表明Nrf2在神经元、星形胶质细胞和胶质细胞中表达(Cuadrado et al., 2019; Shih et al., 2003)。然而,Nrf2在星形胶质细胞中的表达高于神经元。微阵列分析显示Nrf2激活诱导星形胶质细胞中大量Nrf2靶基因(Lee et al., 2003)。在几种神经退行性实验模型中,星形细胞Nrf2表达具有神经保护作用(Cuadrado et al., 2018; Yu et al., 2019)。Nrf2及其驱动基因(NQO1、HO-1和GCLC)的表达在老年人和AD大脑中均出现下降(Silva-Palacios et al., 2018)。Nrf2通过保护性NFE2L2单倍型的高表达可防止神经退行性变。另一方面,Nrf2表达降低与AD风险增加或早期发病相关(von Otter et al., 2010)。Nrf2介导的抗氧化反应的抑制被发现是导致过早衰老表型的关键因素(Kubben et al., 2016)。 Rojo等比较了Nrf2缺乏、衰老和AD小鼠模型之间的差异。他们发现了Nrf2缺乏和衰老之间的7个共同特征,以及Nrf2缺乏和AD之间的10个共同特征。作者还注意到不溶性p-tau蛋白和缺氧小鼠nrf2水平升高(Rojo et al., 2017)。本研究提示Nrf2缺乏与衰老/AD密切相关。这些来自不同实验室的发现支持了Nrf2可能在脑衰老和AD之间起分子连接作用的假设(Branca et al., 2017)。大量证据表明Nrf2功能障碍在认知障碍和AD的发病机制中起作用。Branca等人报道,在AD的APP/PS1小鼠模型中,Nrf2的缺失显著加重了包括空间学习和记忆在内的认知缺陷(Branca et al., 2017)。Nrf2还可以通过上调参与大自噬(Pajares et al., 2016)和伴侣蛋白介导的自噬(Pajares et al., 2018)的基因表达,帮助清除淀粉样蛋白前体蛋白(APP)和tau蛋白。这些发现可能解释了在app损伤和tau损伤的神经元中Nrf2水平的升高,并提示Nrf2激活可能在疾病进展的轻度认知障碍阶段很快发生。最近,Uruno等人证明Nrf2的诱导通过抑制OS和神经炎症改善了AD小鼠模型中的认知障碍(Uruno et al., 2020)。几种Nrf2激活因子在终止AD关键发病机制中也显示出强大的活性,包括抗抑郁药物和p-tau,以及对神经炎症和线粒体功能的调节(Ikram et al., 2019; Yang et al., 2018)。这些结果提示Nrf2是AD重要的治疗靶点。除了已知的Nrf2在维持细胞氧化还原稳态中的作用外,我们还进一步了解了Nrf2与AD关键发病因子的相互作用。
4.1. Nrf2和Aβ
AD的组织学特征是细胞外Aβ斑块沉积(Irvine et al., 2008;Selkoe and Hardy, 2016)。Aβ肽类(~4 kDa)是通过内含体中BACE1和γ-分泌酶顺序裂解APP产生的。BACE1是产生Aβ的必要物质,少量的BACE1的增加会引起一个显著的产量的增加。与分泌酶不同的是,BACE1的水平在AD中被发现升高,这表明它在AD中对细胞积累起重要作用。因此,BACE1的抑制剂正被寻求作为AD的治疗药物。Aβ易于聚集成寡聚体、原纤维和纤维形式的β-折叠构象。Aβ聚集的过程也涉及ROS的产生,导致神经元死亡(Andersen, 2004; Klein et al., 2001)。在AD大脑中发现了几种形式的Aβ,尤其是疏水性较强的Aβ,例如Aβ42,由于其高聚集倾向(Thal et al., 2015)。有证据表明,不同池中的Aβ数量和溶解度可能与疾病状态密切相关(Murphy and LeVine, 2010)。一些靶向聚合体或抑制BACE1的药物正在临床试验中,但它们都没有获得美国食品和药物管理局(FDA)的批准(Dong et al., 2019; Yan and Vassar, 2014)。
APP/PS1小鼠Nrf2基因删除显示炎症反应增强和细胞内APP、β42和β40水平升高。Nrf2缺失的APP/PS1小鼠神经元也显示了核内体、溶酶体和多泡小体的增加(Joshi et al., 2015)。这些发现为Nrf2在APP/ Aβ处理和积累以及自噬功能障碍中的作用提供了线索。Kerr等人首次提供了体内证据,证明通过特异性抑制Keap1激活Nrf2,可以防止AD诱导的Aβ42神经元毒性(Kerr et al., 2017)。Yu等人的一项研究也显示,当他们在AD动物模型中使用Nrf2激活剂isoastilbin时,也有类似的效果,并且他们发现Aβ42和 p tau表达降低(图4) (Yu et al., 2019)。Li等证明西格列汀和槲皮素的结合能够降低大鼠的Aβ表达水平(Li et al., 2019b)。Bahn等发现Nrf2通过与BACE1启动子中的AREs结合抑制了AD动物模型中BACE1的表达。Nrf2的激活减少了BACE1转录和Aβ产物的生成,并改善了认知缺陷。另一方面,Nrf2的缺失增加了BACE1的表达和Aβ生成,并加重了认知障碍。作者还证明,Nrf2对BACE1的调控与ROS抑制无关(Bahn et al., 2019)。本研究不仅将Nrf2的功能与Aβ生成联系起来,还揭示了Nrf2抑制BACE1的能力。有趣的是,该研究还发现Nrf2并不影响细胞分泌酶的活性或表达。这些发现表明,Nrf2的激活不仅抑制了Aβ的产生,还减少了其聚集所造成的损伤。
4.2. Nrf2和p-tau
MAP家族特别是MAP2和tau调节神经元中不同的细胞骨架过程(Amos and Schlieper, 2005)。Tau蛋白是一种55 kDa的蛋白,具有85个以上的翻译后修饰位点,如磷酸化、乙酰化和泛素化。虽然其正常功能需要翻译后磷酸化,但过度磷酸化会破坏其活性(Li and Gotz, 2017; Marciani, 2018; Morishima-Kawashima and Ihara, 2002)。Tau病理的发生是由于tau蛋白的错误折叠导致了β-折叠纤维的产生和积累。Tau病理和β-折叠纤维积累是包括AD在内的几种神经疾病的主要发病机制(Almansoub et al., 2019)。Tau病理被认为是AD认知能力下降和神经退行性变的主要驱动因素(Aschenbrenner et al., 2018; Hanseeuw et al., 2019)。
Jo等研究表明Nrf2可通过诱导核点蛋白52 (nuclear dot protein 52, NDP52)降低p-tau水平。NDP52是一种自噬接头蛋白,其诱导可促进自噬介导的p-tau蛋白降解(图4)。Jo等人也揭示了Nrf2活性的机制,即Nrf2与NDP52启动子中的AREs结合(Jo et al., 2014)。最近,Tang等报道Nrf2调节选择性自噬过程,可促进tau的清除。他们观察到,与年轻的Nrf2−/−动物相比,衰老Nrf2−/−动物中BAG3、NBR1、NDP52和p62的表达显著减少,这表明Nrf2在衰老过程中对维持这些基因的适当表达水平起着关键作用(Tang et al., 2018)。许多Nrf2激活剂,包括萝卜硫素(Kim et al., 2013)、苯福硫胺(Tapias et al., 2018)、富马酸二甲酯(Cuadrado et al., 2018)、大蒜素(Zhu et al., 2015)和mini-GAGR (Murphy et al., 2018)对p-tau AD动物模型显示出积极作用。
4.3. Nrf2 和神经炎症
小胶质细胞是常驻的免疫巨噬细胞样细胞,它们在急性感染和/或毒性损伤时的激活可能有助于清除受损细胞。在AD中,小胶质细胞可能被长期激活,导致有害的促炎细胞因子和ROS/RNS的持续分泌。这些过程导致神经退行性变。神经胶质细胞,位于Aβ斑块区域,表明激活的NF-κB、以及促炎细胞因子和趋化因子水平增加( Yin et al., 2017)。同时, NF-?B被发现调控BACE1表达 (Chen et al., 2012)。Terada等观察到tau病理与AD患者海马旁神经炎症呈正相关(Terada et al., 2019)。提示神经炎症可能在AD的发生发展中起重要作用。因此,神经炎症的消除可以减轻AD的进展和症状(Yang et al., 2019)。Nrf2和NF -κB之间的关系呈现出“阴阳”的概念。尽管在操作系统被激活,Nrf2诱导基因主要是抗炎(包括CD36、IL-17D)而NF –κB作用相反(增加IL-6、COX2和iNOS)。在这种背景下,Nrf2和NF-κB之间的不平衡对AD的发病机制可能是重要的。因此,Nrf2可能在AD中发挥重要的抗神经炎症作用。几项研究证实,Nrf2的激活可以抑制因注射Aβ 或tau引起的小胶质细胞激活,并进一步保护小胶质细胞引发的神经元丢失(Seo et al., 2017; Tom et al., 2019)。
4.4. Nrf2和线粒体功能障碍
线粒体是一种灵活的超微结构细胞器,它控制着关键分子元素的生物能量。线粒体的整体动态本质是由融合和分裂活动控制的。通过抑制线粒体破碎,融合可以延迟细胞凋亡的发生,而分裂对细胞凋亡有积极作用。线粒体质量控制过程中的失衡可能导致线粒体功能障碍(Cheng and Bai, 2018)。在Nrf2敲除小鼠中观察到年龄相关的线粒体OS以及线粒体功能受损,而Nrf2的激活增强了线粒体功能(Kitaoka et al., 2019)。Nrf2激活可增加蛋白酶体活性,而蛋白酶体活性可增强动力蛋白相关蛋白1 (Drp1)的降解(Sabouny et al., 2017)。Drp1是dynamin超家族的一种大型GTPase,它是促凋亡蛋白Bax线粒体转位所必需的酶,并与之结合以增强线粒体外膜通透性。这一过程促进细胞色素c的释放(Wang et al., 2015)。限制线粒体分裂蛋白的水平有利于线粒体低灌注,从而维持线粒体质量控制系统的功能。 Nrf2还控制线粒体抗凋亡蛋白(如Bcl-2和Bcl-xL)的表达、线粒体动力学、包括生物发生,通过与过氧化物酶体增殖激活受体-r 共激活剂 1a (PGC-1a)相互作用,以及通过p62依赖、PINK1/ parkino不依赖机制实现线粒体自噬(East et al., 2014; Merry and Ristow, 2016; Navarro et al., 2017)。AD早期出现线粒体功能障碍,导致神经退行性变,促进突触损伤和细胞凋亡。 、APP、Aβ和p-tau可干扰线粒体成分,导致线粒体动力学异常、生物发生缺陷、自噬减少、线粒体自噬减少、ATP生成受损和氧化损伤增加(Reddy et al., 2018; Sotolongo et al., 2020)。Nrf2可能减少这些损伤并维持线粒体功能(Fu et al., 2018)。Nrf2介导的Drp1降低也可能降低p-tau水平,提高突触活性(Cieri et al., 2018; Kandimalla et al., 2016)。几种Nrf2激活因子能够改善不同AD模型中的线粒体功能障碍(Li et al., 2019a; Liu et al., 2019; Tapias et al., 2018)。Nrf2在线粒体保护中的作用是Nrf2激活在AD治疗中的额外有益作用。
4.5. Nrf2和乙酰胆碱酯酶
乙酰胆碱(ACh)是一种有机神经递质,在神经末梢释放。乙酰胆碱乙酰转移酶(ChAT)以乙酰辅酶A和胆碱为底物在胆碱能神经元中产生乙酰胆碱。乙酰胆碱释放后,通过乙酰胆碱酯酶代谢成胆碱和醋酸盐。在AD患者的大脑中,已经观察到ACh水平的缺乏和AChE水平的增加。乙酰胆碱酯酶通过快速水解乙酰胆碱能突触终止冲动传递。因此,最小化活性乙酰胆碱酯酶水平可以增强胆碱能功能(Osama et al., 2017; Tabet, 2006)。尽管目前尚无明确证据表明Nrf2对乙酰胆碱酯酶(AChE)的影响,但在AD小鼠模型中,几种Nrf2激活因子均对胆碱能功能产生了积极影响,并降低了乙酰胆碱酯酶(AChE)水平,显著提高了乙酰胆碱酯酶(ACh)和乙酰胆碱酯酶(ChAT)水平(Huanga et al., 2019; Li et al., 2019c; Yu et al., 2019)。假设这些作用可能,至少部分是由于Nrf2的抗凋亡作用,可以防止神经元的丢失。Zhang等人发现,在细胞凋亡开始时,在细胞质中发现AChE,然后在细胞核或凋亡小体中发现,并细胞死亡。此外,凋亡细胞中表达的AChE与突触型AChE相同。此外,使用药理抑制剂或反义(阻断乙酰胆碱酯酶的表达)来最小化活性乙酰胆碱酯酶的水平,可以抑制细胞凋亡。他们的结论是,AChE可能是细胞凋亡的标志物和调节因子(Zhang et al., 2002)。这可能解释了AD大脑中大量细胞凋亡导致的高水平疼痛。Nrf2的抗凋亡作用可减少胆碱能神经元的丢失,从而维持乙酰胆碱的产生,使神经认知能力下降最小化。然而,对于AChE在细胞凋亡中的作用以及Nrf2的激活对AChE功能和分布的影响还需要进一步的研究。
5. AD中Nrf2相关通路的研究
一些研究也对AD中的Nrf2相关通路进行了研究,我们在此简要回顾其中一些。p62是一种细胞内信号蛋白,参与多种细胞环境。它还通过自噬参与聚合蛋白的降解。对p62基因敲除小鼠的研究表明,它们表现出一些类似AD的神经退行性变特征。郑等研究了AD模型中p62- Keap1-Nrf2关系,发现AD中p62水平降低可能与Nrf2水平降低有关。作者还推测这可能是AD中tau蛋白过度磷酸化和随后的神经元损伤的原因(Zheng et al., 2012)。Gu等人指出,Nrf2介导的p62信号通路的激活可通过激活自噬来改善由Aβ42损伤引起的细胞死亡(Gu et al., 2018)。Xu等人证实了p62在自噬介导的对致病性微管相关蛋白tau (MAPT)的清除中发挥的作用,并通过MAPT阻断了神经纤维缠结的积累和病理扩散。他们还表明,p62介导的对不溶性突变体MAPT的清除是非泛素依赖的(Xu et al., 2019)。
多项研究表明,AD中GSK-3β水平升高(Leroy et al., 2007; Ochalek et al., 2017; Pei et al., 1997; Virpi Talman et al., 2016)。GSK-3β抑制蛋白活性的增加直接参与了磷酸化导向的Nrf2的降解(图4)。GSK-3β抑制蛋白的基因过表达导致神经元丢失和记忆缺失,并参与了小鼠tau蛋白的过度磷酸化(Engel et al., 2006; Hernandez ´ et al., 2002)。Tideglusib是GSK-3β抑制因子,能够通过最小化tau蛋白磷酸化和减少淀粉样蛋白沉积来改善认知(Sereno et al., 2009)。Cuadrado等研究表明,富马酸二甲酯可以抑制Keap1和GSK-3β细胞,使Nrf2发生两层上调。在动物模型中,富马酸二甲酯能够显著降低tau蛋白磷酸化,并提供抗tau病理的保护(Cuadrado et al., 2018)。这一发现鼓励了对这类化合物的研究,它们可能对神经退行性疾病的治疗有用。
p38 MAPK在AD中被证实上调(Sheng et al., 2001; Sun, 2003)。除了降低Nrf2核易位外,它还可能通过OS激活Aβ(Giraldo et al., 2014)。最近的研究表明,p38的减少和Nrf2水平的升高可以消除SH-SY5Y细胞的神经毒性和OS (Amin et al., 2017a; Bach et al., 2011)。AD中NF-κB-p65的水平增加 (Terai et al ., 1996)。几个不同的AD模型研究表明,抑制NF-κB可以调控的表达Nrf2(Zhao et al., 2018)。Tom等人表明gedunin, Nrf2活化剂,可以通过抑制NF-κB及减少神经炎症,保护细胞免受Aβ42 寡聚体诱导的神经毒性 (Tom et al., 2019)。
6. Nrf2激活剂用于AD临床试验的影响
自1906年AD出现以来,只有五种药物被FDA批准用于临床治疗。这五种药是他克林,利瓦斯明,加兰他敏,多奈哌齐和美金刚胺。前四种药物是乙酰胆碱酯酶抑制剂,可以改善认知功能。美金刚胺是一种n -甲基- d -天冬氨酸(NMDA)受体拮抗剂,常用于治疗中重度AD。然而,这些药物都不能终止或逆转AD的潜在进展。它们只能暂时改善认知功能。他克林、利瓦斯明、加兰他明和多奈哌齐会产生一系列副作用,包括呕吐、腹泻、恶心、头晕和头痛。不幸的是,到目前为止,在AD的临床试验中,没有一种影响到Aβ和/或p-tau的分子显示出显著的作用(Dong et al., 2019)。所有这些因素使得新的治疗策略更具吸引力。如上所述,Nrf2可以通过直接和间接的方式干预Aβ和p-tau。此外,大多数AD患者是老年人,因此他们也可能患有与AD有共同病理特征的其他年龄相关疾病。Nrf2激活被发现对几种与年龄相关的疾病有用(Deshmukh et al., 2017; Francisqueti-Ferron et al., 2019; Silva-Palacios et al., 2018)。多靶点治疗被提出,因为乙酰胆碱酯酶抑制剂与抗氧化剂或NMDA受体拮抗剂一起给予治疗效果更好(Mangialasche et al., 2010)。但在这种情况下,使用单一的Nrf2激活剂可能对几种疾病有益,因此可能降低成本、副作用和药物相互作用。许多天然和合成的Nrf2激活剂已在神经退行性疾病的临床前研究或模型中得到验证(Dinkova-Kostova et al., 2017; Hou et al., 2019; Hou et al., 2018; Peng et al., 2019a; Peng et al., 2019b; Peng et al., 2015c; Yao et al., 2015)。最近,一些纳入AD临床试验的药物被证实能够激活Nrf2(表1)。2014年,苯磷硫胺被纳入AD临床试验(NCT02292238)。4年后,Tapias等人在体内模型中表明,苯磷硫胺可以激活Nrf2,并降低P301S AD中的p-tau (Tapias et al., 2018)。同样,DL-3-n-丁基苯酞(NCT02711683) (Qi et al., 2018)和培哚普利 (NCT02085265) (Kamel et al., 2020)的Nrf2激活作用最近也得到了验证。从十字花科植物中分离得到的萝卜硫素进入AD临床试验(NCT04213391)可能是开发基于Nrf2激活的AD新药的一个很好的步骤。萝卜硫素通过与Keap1的Cys151相互作用,也是Nrf2亲电激活剂。萝卜硫素具有神经保护作用,干预Aβ 和 p tau,并改善急性AD的认知缺陷(Kim et al., 2013)。目前,萝卜硫素有20多个临床试验。然而,萝卜硫素在室温下相对不稳定。因此,它的合成类似物SFX-01 (Evgen Pharma开发的药物)备受关注。这些能力使萝卜硫素及其类似物成为AD药物开发的重要药物。
在FDA批准的可激活Nrf2的药物中,富马酸二甲酯被认为是治疗AD的有前景的药物。2013年,富马酸二甲酯被FDA批准用于治疗多发性硬化症。富马酸二甲酯在不同的AD模型中均表现出积极的作用,因此被认为是药物重新定位的最佳选择之一。总的来说,半不饱和羰基化合物是研究最多的可激活Nrf2的化合物。这类化合物被设计通过与Keap1的半胱氨酸残基结合来干扰Keap1- nrf2的相互作用(Peng et al., 2015b)。几种自然产生的药剂,包括xanthohumol, 6 dehydrogingerdione, cardamonin, 6-shogaol and chlorogenic acid,属于这一家族,并已被我们实验室鉴定为通过激活Nrf2来发挥神经保护能力(Bai et al., 2019; Peng et al., 2017; Peng et al., 2015a; Yao et al., 2014; Yao et al., 2019)。
目前,有数十种天然和合成的Nrf2激活剂可用于进一步深入研究其在AD模型中的作用。表2总结了参与不同临床试验并证明对AD动物模型有积极影响的化合物。这些化合物可能是药物开发的良好起点。化学修饰也可能有助于改善这些分子的化学和药理特性,从而达到更好的治疗效果。
7. Nrf2治疗AD药物开发的挑战
找到一种有效且相对安全的治疗AD的新药不是一件容易的事。此外,通过靶向Nrf2探索这种新的治疗方法也使这项任务更具挑战性。在这里,我们讨论几个问题和考虑开发Nrf2激活剂的临床应用。
7.1. 选择性
Nrf2抑制蛋白Keap1是一个富含Cys的蛋白,有27个Cys残基,其中一些高度反应。Keap1的这一特性促使许多研究小组设计能够与Keap1的Cys残基硫醇基团形成共价键的共价抑制剂。在27个Cys残基中,Cys151、Cys273和Cys288得到了高度关注。Cys273和Cys288在基础和应激条件下Nrf2的调控中都发挥了重要作用。另一方面,Cys151在应对各种应激条件方面具有重要作用(Suzuki et al., 2013)。许多亲电物种能够与Keap1的Cys残基形成共价相互作用,从而抑制Nrf2泛素化。用亲电修饰剂随机或选择性地攻击Keap1 Cys残基被称为“半胱氨酸编码”(Cuadrado et al., 2019; Fao et al., 2019)。不幸的是,攻击Keap1中的Cys残基并不意味着不能攻击其他富含半胱氨酸的蛋白质。这种缺乏选择性是开发共价Keap1修饰剂的主要问题(Suzuki et al., 2013)。人们正在努力发现更有选择性的化合物,蛋白质-蛋白质相互作用(PPI)抑制剂也在选择之列。CPUY192018是通过阻断Nrf2和Keap1的PPI来激活Nrf2的最成功的例子之一(Lu et al., 2019)。基于更大的支架结构可能提供更好的选择性的假设,也研究了五环迈克尔受体的氰烯酮三萜。Bardoxolone甲基就是这类化合物的一部分,目前正在进行四个临床试验(Cuadrado et al., 2019)。Nrf2可以通过其他不同的策略激活,从而提高选择性。Nrf2激活的途径之一是使用竞争性抑制剂攻击Keap1-Nrf2的结合位点。这一策略是实用的和高度选择性的,因此它可能是减少副作用的一个很好的替代方法(Madden and Itzhaki, 2020)。另一种方法是靶向BTB结构域中的Keap1二聚作用(Zhuang et al., 2009)。BTB结构域也参与了Keap1和Cul3的结合,针对这种Keap1-Cul3的相互作用也可能导致Nrf2的激活。Keap1 IVR域中发现的靶向3-box也可能干扰Keap1和Cul3的结合(Cleasby et al., 2014)。用新的激活剂靶向这些不同的位点可以增加选择性,从而减少潜在药物的副作用。此外,靶向激活AD脑损伤区域的Nrf2是另一个挑战,进一步修饰这些化合物可能是提高选择性的一个有希望的策略。
7.2. AD实验模型
在药物发现领域,实验模型是非常重要的。这些模型在阐明疾病的病理生理原因和测试候选药物的临床前后果方面至关重要。在AD的案例中,对多个物种的神经病理学调查显示,AD似乎是人类特有的(Drummond and Wisniewski, 2016)。对包括恒河猴、狒狒、黑猩猩、食蟹猕猴和狐猴在内的非人类模型的研究表明,它们具有AD的一些共同特征,有助于解开AD病理的一些模糊之处。不幸的是,在这些模型上进行研究可能会花费很长时间,毕竟,它们可能会随着它们表现出的特征而变化,因为它们没有受到人类AD的折磨。AD治疗学的临床试验失败率极高(>99%),这可能与缺乏合适的AD动物模型有关。在人工诱导非人类AD模型方面已经做了很多工作,包括注射模型和转基因小鼠模型。这些模型可用于数百种潜在药物的AD临床前测试(Esquerda-Canals et al., 2017)。不幸的是,在数百种临床前成功的候选药物中,只有5种获得了市场批准。在临床试验中未能取得成功可能是因为这些模型考虑了毒性APP和/或tau突变蛋白的表达,而忽略了其他与疾病相关的下降,如与Nrf2相关的稳态功能。虽然Nrf2敲除小鼠表现出与衰老和AD共同的特征(Rojo et al., 2017),但该模型是否与AD病理更相关还需要更多的努力来验证。
7.3. 能否通过血脑屏障(BBB)
为了达到预期的药理学效果,需要考虑许多候选药物的因素,包括生物利用度、代谢稳定性、溶解性和细胞通透性。此外,药物应表现出更多的亲和力,或至少可以达到目标器官的足够剂量(Krogsgaard-Larsen et al., 2005)。因此,要激活AD神经元中的Nrf2,药物必须以足够的浓度到达AD脑损伤区,使其能够发挥作用。血脑屏障是保护中枢神经系统的天然屏障,通过阻止神经毒性血浆物种、血细胞和病原体进入大脑。不能跨越血脑屏障的药物获得AD临床试验成功的机会很低(Pardridge, 2019)。细胞培养研究可能大大高估了测试药物的活性。由于生物利用度低,这些候选药物在临床试验中可能表现出令人失望的结果。分子量< 450道尔顿、极性表面积< 90 A2和氢键供体数< 3的药物更有可能穿透血BBB (Hanson and Frey, 2008)。这些通用指南为中枢神经系统药物的设计和开发提供了初步考虑。然而,血脑屏障的侵彻能力必须通过实验来评估。因此,需要在亲脂性、分子构型和分子大小之间实现神奇的结合和平衡,才能使药物通过血脑屏障。
7.4. 确定给药方案
关于Nrf2激活治疗AD的另一个考虑是,老年人和不太健康的受试者对Nrf2激活剂的反应降低(Hammer et al., 2018)。因此,给药剂量必须考虑患者的年龄和健康状况。由于Nrf2与其靶基因相比半衰期较短,因此也应通过分析药物分布和Nrf2基因在可接近细胞或组织中的表达特征来间接推断合适的给药方案(Cuadrado et al., 2019)。
8. 结论
Nrf2是抗氧化系统的主要控制器,负责II期相解毒酶和抗氧化蛋白的组成性表达。大量的生物分子参与了Nrf2的调控,这说明了这一途径的复杂性。AD中Nrf2、p62 GSK-3β,p38和NF -κB-p65水平发生改变。大量证据表明,Nrf2具有在各种AD模型中降低OS、炎症和线粒体功能障碍的能力(图5)。有趣的是,Nrf2也可以抑制BACE1的表达,并改善Aβ介导的毒性。Nrf2还能降低AD模型中p-tau和认知缺陷的水平。几种Nrf2激活剂在不同的AD动物模型中显示了良好的效果。基于这些研究,以及并没有FDA批准新的药物用于临床治疗AD, Nrf2激活似乎是一种新颖而有吸引力的治疗方法。然而,我们并不认为这种治疗方法会比其他方法更容易。Nrf2通路本身复杂,可通过不同的机制激活。此外,Nrf2激活剂的开发也面临着许多挑战。用亲电共价抑制剂攻击Keap1中的Cys残基是Nrf2激活的最常见机制。不幸的是,这种类型的活化剂通常缺乏选择性。低选择性药物有望引起广泛的副作用。有几种方法可以用来解决这个问题,包括开发PPI抑制剂和非共价Keap1抑制剂。缺乏理想的AD动物模型是AD药物发现领域的另一个障碍。到目前为止,关于Nrf2激活在人类AD大脑中的作用的信息很少。最近在AD临床试验中加入的Nrf2激活因子,如萝卜硫素,可能为Nrf2激活在AD中的结果提供有价值的信息。
图1:AD大脑遭受氧化应激。(a)线粒体功能障碍可能导致ROS产生增加和细胞凋亡。(b)在AD中观察到广泛的脂质过氧化。(c) AD大脑还显示出神经毒性微量元素如铁、铝、铜和汞的水平提高,这些元素可诱导自由基的形成(铁就是一个例子)。(d)高水平Aβ(Aβ42;PDB: 5kk3)也可促进AD脑内的氧化应激。(e)高水平的ROS可能攻击DNA,因此AD脑显示更多的氧化DNA(鸟嘌呤为例)。
图2:Nrf2和Keap1蛋白不同结构域的示意图。(a) Nrf2的七个Neh域及其功能。(b) Keap1的五个代表性区域和几个重要的半胱氨酸传感器在不同区域发现。
图3:Nrf2的keap1依赖性和非依赖调控机制。在基础条件下,Keap1-Cul3复合物与Nrf2结合并介导其泛素化和降解。p62可能通过与Keap1结合,导致Keap1的自噬降解,从而减少了这一过程,从而增加了细胞核中Nrf2的积累。GSK-3β和p38 MAPK可使Nrf2在Neh6区域磷酸化并有利于其降解。相反,PKC、CK2 PERK、JNK1和ERK2介导的Nrf2磷酸化促进了Nrf2与Keap1的分离和核积累。在细胞核中,Nrf2与转录共激活因子Mafs形成异源二聚体,促进与ARE的稳定结合并增强基因转录。Fyn激酶可能使Nrf2在Tyr568处磷酸化,有利于其核出口。Bach1与Nrf2竞争结合导致Nrf2转录能力下降。NF-κB也通过与ARE结合与Nrf2竞争, 下调其转录活性。
图4:AD中Nrf2与一个高亮区域(蓝色)和p-tau(灰色)相互作用的示意图。Aβ通过内含体中BACE1和γ-分泌酶的蛋白水解功能从APP中衍生出来。Aβ分泌入组织间液,可导致Nrf2水平降低。Aβ可聚集成寡聚体和原纤维,对细胞功能有许多影响,包括突触活性受损和脑毛细血管血流量受损。Nrf2的激活降低了BACE1的转录水平,从而降低了Aβ的产生。Nrf2还可能降低聚合Aβ的水平,从而减少AD淀粉样蛋白的病理。错误折叠的MAPT和p-tau蛋白的积累可能导致成对螺旋丝的形成(PHF;PDB: 6HRE),随后跨突触传播到远处的神经元。Nrf2的激活可能会增加参与选择性自噬过程的p62和NDP52的水平。P62还可以促进不溶性MAPT的降解,从而降低神经纤维缠结(NFTs)水平。Nrf2可能预防p-tau诱导的神经退行性变,也可能减少p-tau的聚集。因此,Nrf2的激活导致AD tau的病理减少。
图5:Nrf2在AD中的作用的简化说明。Nrf2通过(上调/下调)调控多个下游靶点来抑制AD的几个关键致病特征。
本文由福山生物整理翻译,转载请注明出处。