






最新资讯



-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
阿尔茨海默病治疗的新靶点:Nrf2通路与自噬之间的串扰
发表于:2021-12-14 作者:admin 来源:本站 点击量:16233
原文:Zhang W , Feng C , Jiang H . Novel Target for Treating Alzheimer's Diseases: Crosstalk between the Nrf2 Pathway and Autophagy[J]. Ageing Research Reviews, 2020, 65(1):101207.
翻译:
亮点:
• p62连接Nrf2通路和自噬以维持体内稳态。
• Nrf2的激活和自噬有利于AD治疗。
• p62-Keap1-Nrf2轴是治疗AD的新型潜在机制。
• 激活p62-Keap1-Nrf2轴的天然化合物是治疗AD的有效药物。
摘要:
在哺乳动物中,Keap1-Nrf2-ARE途径(以下简称“ Nrf2途径”)和自噬是主要的细胞内防御系统,可对抗氧化损伤并维持体内平衡。p62 / SQSTM1是一种泛素结合的自噬受体蛋白,它连接Nrf2途径和自噬。p62的磷酸化显著增强了其对Keap1的亲和力,从而诱导Keap1释放Nrf2,而p62-Keap1异二聚体募集了LC3并介导了Keap1在选择性自噬途径中的永久降解。最终,Nrf2积累在细胞质中,然后转移到细胞核中以激活下游编码抗氧化酶基因的转录,从而保护细胞免受氧化损伤。由于Nrf2也上调了p62基因的表达,因此创建了p62-Keap1-Nrf2正反馈环,该环进一步增强了对细胞的保护作用。研究表明,p62激活的非经典Nrf2途径是神经变性疾病的重要标志。p62-Keap1-Nrf2正反馈回路和Nrf2通路参与消除AD诱导的ROS和蛋白质聚集。因此,维持p62Keap1-Nrf2正反馈环路(是Nrf2途径与自噬之间的桥梁)的体内平衡可能是治疗AD的潜在目标。
关键词:Nrf2通路、自噬、氧化应激、p62-Keap1-Nrf2正反馈回路、阿尔茨海默病
1. 前言
活性氧(ROS)是正常有氧代谢的副产物,在多种危险因素中产生,如环境应激、衰老和疾病。高水平的ROS破坏DNA结构和膜分布,改变蛋白质结构和功能,导致细胞损伤和死亡。氧化应激反应清除体内过多的ROS,维持体内氧化还原稳态(Yi et al., 2020; Xia et al., 2020)。核因子、红细胞来源因子2相关因子(Nrf2)是抗氧化防御的关键转录因子,与Kelch like ech相关蛋白1 (Keap1)相互作用。在氧化应激条件下,由于过多的活性氧产量,Nrf2 Keap1分离,进入细胞核,它结合抗氧化反应的元素(ARE)触发下游抗氧化酶的表达,如血红素加氧酶1 (HO1)、超氧化物歧化酶(SOD)、过氧化氢酶(CAT)和NAD (P) H醌脱氢酶1 (NQO1)。
自噬在真核细胞中广泛存在,是一种自我修复的生物过程,参与细胞生长、代谢和抗氧化应激。自噬负责清除长寿命或错误折叠的蛋白质、脂滴和受损的细胞器,以保持细胞内稳态(Liu et al., 2020)。自噬是一种溶酶体依赖的自我降解途径。根据底物运输到溶酶体进行降解的方式,自噬途径通常分为三种不同类型,这三种类型分别是微自噬、伴侣介导的自噬(CMA,只发生在哺乳动物)和巨自噬(Suzuki et al., 2017)。在微自噬过程中,底物被运输到溶酶体中,并被小的溶酶体膜的内陷所隔离,这些内陷被挤压到内腔中(Li and Hochstrasser, 2020)。在CMA中,五肽基序(KFERQ)暴露的错误折叠蛋白被热休克70 kDa蛋白8 (HSPA8/HSC70)识别,进而与溶酶体相关膜蛋白2A (LAMP2A)结合。这些蛋白在溶酶体腔内展开和降解(Dou et al., 2020)。自噬最典型的形式是巨自噬(以下简称自噬)。自噬的特点是胞质成分包裹在称为自噬小体的双层膜结构中。然后,自噬体与溶酶体膜融合并被水解酶降解。在营养缺乏的情况下,该途径被激活以提供氨基酸和ATP (Yang et al., 2019)。
p62转录被氧化应激、代谢紊乱、疾病和自噬受阻激活,导致细胞质中大量p62聚集。p62的聚集物通过与Nrf2抑制蛋白Keap1直接相互作用,维持Nrf2的慢性激活,并且p62介导Keap1在选择性自噬途径中的永久降解(Sanchez-Martin and Komatsu, 2018)。因此,Nrf2信号通路被激活,导致编码抗氧化酶的基因转录上调(Deng et al., 2020)。此外,核内Nrf2促进p62基因的过表达,形成p62- keap1 -Nrf2正反馈轴,导致Nrf2的持续激活。与经典途径相比,p62介导的选择性自噬调节Nrf2信号通路属于非经典机制。越来越多的研究表明,p62-Keap1-Nrf2通路的失调与神经退行性疾病的治疗有关(Shah et al., 2018)。因此,本综述将阐述Nrf2通路、自噬及其串扰在AD治疗中的作用。
2. Nrf2 通路
2.1 Keap1的基本结构
Keap1的分子量为66 kDa,由624个氨基酸组成。它是Cullin3 (Cul3)-E3泛素连接酶复合物的底物蛋白,是一种负向调节Nrf2活性的胞质蛋白(Davies et al., 2016)。从结构上看,Keap1由5个功能区组成。第一个区域是N端区域(NTR)。第二个区域是间质区(IVR),其中含有大量的半胱氨酸残基。第三个区域是brac -a-brac (BTB)区域,包括broad complex、tramtrack和brac,其中包括一个重要的应力敏感位点半胱氨酸151 (Cys151)。后两个区域是双甘氨酸重复或Keleh重复(DGR)区域和C末端区域(CTR) (Dinkovakostova et al., 2017; Deshmukh et al., 2017)。在这些区域中,BTB、IVR和Keleh/DGR域是Keap1的重要功能域。BTB结构域是蛋白质-蛋白质相互作用中一个进化保守的序列。这个结构域可以与另一个Keap1分子的BTB区域结合,形成一个同分二聚体,然后与Nrf2结合。该区域还可以与Cul3结合形成E3 Ub连接酶复合物,导致泛素介导的Nrf2降解(Kansanen et al., 2012; Tao et al., 2017)。在BTB结构域,当半胱氨酸残基的结构发生改变时,Nrf2蛋白的泛素化和降解将受到抑制。这一过程诱导Nrf2与Keap1分离并与ARE结合以激活编码抗氧化酶的基因转录。IVR区域富含半胱氨酸残基,对氧化应激敏感,也与Nrf2的稳定性有关。在正常生理条件下,IVR区域参与泛素介导的Nrf2降解。值得一提的是,Cys273和Cys288在抑制Nrf2活性方面发挥了重要作用。当机体被亲电体和ROS激活时,IVR结构域发生转变,导致Nrf2和Keap1相互分离(Magesh et al., 2012)。DGR结构域(包括6个Kelch重复)和CTR区域统称为DC结构域,是Keap1与细胞质肌动蛋白的结合位点。Keap1同型二聚体中的DGR结构域与Nrf2中Neh2结构域的DLG和ETGE氨基酸基序相互作用,对Nrf2活性产生负调控作用。在外源性或内源性刺激下,通过半胱氨酸失活诱导Keap1 DGR区域构象变化,诱导Keap1-Nrf2蛋白复合物解离,释放Nrf2进入细胞核。Keap1的基本结构如图1a所示。
2.2 Nrf2的基本结构
Nrf2分子量为100kda,由624个氨基酸组成。Nrf2属于碱性亮氨酸拉链(basic leucine zipper, bZIP)转录因子家族,其成员保持cap-n-collar (CNC)结构,而人类Nfe2l2位于2q31上(Tavakkoli et al., 2019; Bottino-Rojas et al., 2018)。根据其生理功能,Nrf2可分为7个高度保守的ECH同源结构域(Nrf2- ECH同源),包括Neh1到Neh7 (ECH,与鸡Nrf2具有CNC同源性的红血球来源蛋白)。亮氨酸CNC-bZIP区域位于Neh1结构域,可与细胞核中的一种小的肌肉筋膜性纤维肉瘤(Maf)蛋白(即脊椎动物中的MafF、MafG或MafK)结合,并有助于Nrf2与DNA中的ARE结合。随后,抗氧化酶、Ub酶、II期解毒酶和蛋白酶体的转录上调,以抵御细胞中的氧化应激(Karan et al., 2020; Shen et al., 2019)。Neh2结构域是一个由16个氨基酸组成的短肽,其氨基酸序列在69~84之间,位于Nrf2的N端。Nrf2的Neh2结构域包含分别与Keap1具有高亲和力和低亲和力的ETGE和DLG基序;该区域介导Nrf2的降解并负调控Nrf2的转录活性(Tian et al., 2018)。Nrf2 C -末端的Neh3区域是一个可以结合CHD6并激活ARE的重要功能区域(Zhang et al., 2014)。Neh4和Neh5域可以结合激活cAMP反应元素结合蛋白(CREB)协助Nrf2核转运和以Nrf2-Maf的形式绑定到ARE (Krajka Ku?niak et al ., 2016)。Neh6结构域具有大量的丝氨酸残基(Chen et al., 2020),对Nrf2转录有负向影响,且不依赖于Keap1 (Chowdhry et al., 2013),该结构域主要参与Nrf2的降解。其调控机制是DSGIS和DSAPGS的氨基酸基序可通过β-转介含重复蛋白(β-TrCP)识别,并可诱导Skp1-Cul1-Rbx1/Roc1-β-TrCP Ub连接酶复合物调控Nrf2泛素化介导的降解(Cuadrado, 2015)。Neh7结构域识别视黄素X受体(RXR) (Li et al., 2017)。此外,Nrf2的第40个氨基酸是一个磷酸化位点,该位点的磷酸化导致Nrf2从Keap1中解离。Nrf2的基本结构如图1b所示。
2.3 ARE的基本结构
ARE位于一些细胞保护基因(抗氧化和自噬相关基因)启动子区域上游5'端,属于特定的DNA启动子联合序列(Jeddi et al., 2017)。ARE被定义为顺式作用的DNA增强因子,负责触发氧化应激反应,以诱导II期解毒酶和抗氧化酶的表达。ARE广泛表达于生物体中,其功能是减轻细胞或组织的各种损伤,维持体内平衡。虽然在不同的细胞中ARE略有不同,但共同的核苷酸序列为5'- (G/A) TGA (G/C) XXX GC (G/A) -3' (x代表任何核苷酸)。ARE的基本结构如图1c所示。
2.4 Nrf2信号通路的调控机制
在漫长的进化过程中,细胞获得了抵抗外部毒素和内部代谢毒素的能力。最重要的转录因子Nrf2调节抗氧化应激反应(Erukainure et al., 2020)[29]。Keap1-Nrf2- ARE通路是激活Nrf2的典型调控通路,Nrf2的活性主要由Keap1调控。在基础条件下,Keap1的BTB结构域与Cul3 E3 Ub连接酶复合物结合,两个Keap1分子通过BTB区域形成一个同源二聚体。Keap1-Keap1复合物通过Keap1的DGR域与Nrf2的ETGE和DLG基序以1:1的比例直接结合到Nrf2上(Raghunath et al., 2019)。Nrf2 Neh6域的DSGIS和DSAPGS氨基酸基序与E3 Ub连接酶结合,然后, Ub-蛋白酶体上得Ub转移到Nrf2ETGE、DLG基序之间的赖氨酸残基诱导泛素介导的Nrf2降解,因此保持适当的浓度Nrf2(Lee and Ryu, 2017)。
在氧化应激条件下,线粒体产生大量的高活性分子,如ROS (Chen et al., 2020);当ROS含量超过细胞清除能力时,氧化还原系统出现失衡,导致脂质、蛋白质和DNA发生氧化损伤,最终导致细胞凋亡、组织或器官损伤(Liu et al., 2017; Valavanidis et al., 2013)。因此Nrf2通路被显著激活,具体机制如下。一方面,Keap1 IVR区Cys273和Cys288的巯基被氧化形成二硫键(Magesh et al., 2012),导致空间构象的改变。然后,具有较强结合能力的高亲和力ETGE 基序仍然能够与Keap1紧密结合,而较弱、低亲和力的DLG 基序与Keap1分离。Nrf2的空间定位发生改变,不能被泛素蛋白酶体降解。另一方面,Keap1 BTB结构域的Cys151被修饰,产生空间位阻效应,导致Keap1和Cul3解离(Wang et al., 2020)。这破坏了keap1-cul3 E3 泛素连接酶的活性,并抑制了泛素介导的Nrf2降解。当Keap1-Keap1-Nrf2的数量达到饱和时,Keap1同型二聚体在细胞中不能再生,新生成的Nrf2由于自身的保护,不再与Keap1结合。Nrf2迁移到细胞核并与小Maf蛋白结合。然后,Nrf2-Maf与ARE结合,增加抗氧化蛋白和II期解毒酶的转录激活(Otsuki and Yamamoto, 2019)。当氧化还原平衡恢复后,Nrf2从细胞核转位到细胞质,并通过泛素化降解(Pandey et al., 2017)。Nrf2通路的具体调控机制如图2所示。
3. 自噬的调节机制
从机制上看,自噬主要分为四个阶段,即自噬的启动和诱导、自噬体的扩展和形成、自噬体与溶酶体融合和降解以及降解产物的循环利用(Carlsson and Simonsen, 2015)。在营养充足的条件下,哺乳动物雷帕霉素靶蛋白(mTOR)是自噬的重要调节因子,其作用是抑制自噬。mTOR存在于细胞内的两个复合物中;这些复合物是mTOR复合物1 (mTORC1)和mTOR复合物2 (mTORC2)。mTORC1复合物由mTOR、raptor和富含脯氨酸的Akt底物(PRAS40)组成,raptor是mTORC1激活的重要因素。此外,mTOR、rictor、哺乳动物应激激活的MAP激酶相互作用蛋白(mSIN1)以及与rictor 1和2观察到的蛋白形成mTORC2复合物。unc -51样激酶1 (ULK1)是自噬核心机制中最上游的激酶,ULK1的磷酸化是自噬激活的重要决定因素。AMP活化蛋白激酶(AMPK)和mTORC1可以直接调节ULK1 (Atg1 homologs)的磷酸化,这些蛋白是自噬的重要调节蛋白。AMPK正调控自噬,mTORC1通过与ULK1结合抑制ULK1复合物的磷酸化,从而抑制自噬(Noda, 2017)。
3.1自噬的启动和诱导
正常情况下,自噬被mTORC1抑制,Beclin1与Beclin2在内质网(ER)上形成复合物。在缺乏营养或生长因子的条件下,自噬被认为是非选择性的,并集中于增加任何胞质蛋白和其他大分子的降解,以提供必需的营养物质。在应激条件下,自噬被认为是选择性的,有目的地识别底物并将其紧密地结合到新出现的自噬体膜上。相关机制如下。mTORC1失活后,由ULK1、ULK2、Atg13、Atg101和局灶粘附激酶家族相互作用蛋白(FIP200) (Neufeld, 2020)组成的ULK1复合物被AMPK激活后转运到ER (Chen et al, 2020)。磷酸化的ULK1复合物调控JNK1酶诱导Beclin2的磷酸化,从而改变Beclin2的空间构像,将Beclin1释放到细胞质中(Xue et al., 2015)。Beclin1对应激高度敏感(Kiruthiga et al., 2020),并与III类磷脂酰肌醇3激酶(IIIPI3K/PIK3C3;酵母中的Vps34), Atg14L(也称为BARKOR;酵母中的Atg14), p150(又称PIK3R4;酵母菌Vps15)和AMBRA1。PI3K (PI3P)的磷酸化参与了自噬的启动(Nascimbeni et al., 2017)。部分PI3P复合物征募双FYVE-containing protein 1 (DECP1),被ER包裹以增强膜的弯曲度,并形成一个收集自噬相关蛋白的平台。PI3P与WD重复域磷酸肌醇肽相互作用的另一部分(WIPI, Atg18同源物)在胞浆-液泡(Cvt)通路中发挥重要作用。这个阶段是自噬前体膜的形成(Cao et al., 2019;Nakatogawa, 2020)。
3.2自噬体的延伸和形成
自噬前体的双层膜被不断拉长以形成自噬体。在延长阶段需要两个Atg接合系统被ub -蛋白酶体修饰。一个系统是Atg12-Atg5-Atg16L共轭系统,锚定自噬体的外膜,另一个系统是Atg8/LC3共轭系统,同时连接内膜和外膜。Atg12-Atg5-Atg16L复合物是自噬体形成所必需的。自噬激活后,ub样蛋白Atg12被e1样ub激活酶和e2样ub结合酶诱导,通过Atg7和Atg10作用与Atg5结合。Atg16的低聚物与Atg12-Atg5的共轭物结合,形成位于外膜的Atg12-Atg5- atg16l复合物(Liet al., 2020)。在非自噬条件下,LC3(在酵母中也被称为Atg8同源蛋白)以proLC3的非活性形式存在。自噬激活后,LC3通过Atg4、Atg7、Atg3的顺序作用转化为LC3I。然后Atg12-Atg5-Atg16L复合物介导LC3I与磷脂酰乙醇胺(PE)结合,形成具有膜结合能力的活性蛋白LC3II。LC3II与p62的LIR结构域相互作用,影响扩张吞噬团的曲率和自噬前体的形成。自噬前体膜通过诱导LC3II不断延伸形成囊状自噬体,嵌入双层膜表面(Noda and Inagaki, 2015)。
3.3自噬体与溶酶体的融合和产物降解
自噬体与溶酶体的融合是通过两种机制:一个机制是直接的自噬体与溶酶体融合形成自吞噬泡,和其他机制的结合与成熟的核内体自噬体形成自噬核内体,后来搬到溶酶体与溶酶体融合。细胞核周围的微管组织中心(MTOC)含有大量的溶酶体。在压力作用下,膜附LC3-II蛋白在肌动蛋白复合物的作用下移动到MTOC附近。同时,溶酶体向MTOC移动,加速了相互融合的效率。在自噬溶酶体形成初期,ATP为溶酶体膜上的质子泵提供能量,增加溶酶体主动摄取H+。阳离子和阴离子泵可以维持溶酶体中的离子电流平衡(如Ca2+、K+和Cl-)。当内溶酶体的pH值在4.5 ~ 5.0时,水解酶被激活,困在自噬体内的LC3II蛋白与底物一起最终被降解。自噬体外膜上LC3-II被Atg4降解回收。基于多糖的膜蛋白(如LAMP1和LAMP2)嵌入溶酶体膜表面,可以防止溶酶体自身降解。自噬溶酶体融合后,水解酶降解底物,包括脂肪酸和氨基酸在内的水解物参与细胞内循环,提供能量(Zhao et al., 2020)。具体流程如图3所示。
4. 细胞中的p62-Keap1-Nrf2轴
4.1 p62的基本结构
p62,又称SQSTM1,是一种与ub结合的自噬受体,分子量为62 kDa。p62主要位于细胞质中,可在细胞质与细胞核之间转运。p62含有440个氨基酸,由9功能域:phox bem1域(PB1), ZZ-type锌指域(ZZ) TRAF6绑定域(TB),核本地化信号(NLS),核出口信号(NES), PEST域,LC3交互领域(LIR), Keap1相互作用域(KIR)和C末端 Ub相关(UBA)域(Lin et al., 2013)。p62通过PB1结构域的selfoligomerization形成聚合体,该结构域还与PKC、胞外信号调节激酶5 (MEK5)和邻近的BRCA1基因1蛋白(NBR1)相互作用,形成异源二聚体。c端UBA结构域,对Ub具有高亲和力的高度保守序列(Katsuragi et al., 2015; Denk et al., 2019),对于隔离转运到自噬体中的泛素化底物至关重要。此外,UBA结构域的S403和S407可被靶向形成Ub链48K的蛋白聚合物(Zhang et al., 2019),并被由两个亚基组成的26S蛋白酶体识别,这两个亚基是一个20S蛋白酶体和一个19S调节粒子。19S粒子识别泛素化的p62蛋白,并通过狭窄的孔将其转移至20S蛋白酶体,20S蛋白酶体将底物消化为包含2-24个氨基酸的肽,并为细胞再利用提供必要的材料(Jakobi et al., 2020; Cohen-Kaplan et al., 2017)。ZZ域与受体相互作用蛋白1 (RIP1)调节肿瘤坏死通路和NF-κB信号(Yan et al ., 2017)。TB结构域结合TRAF6催化赖氨酸(K)残基位点(63位)多聚泛素链的形成(Kim et al., 2019)。ZZ和TB区域之间的未知区域与raptor相互作用,激活雷帕霉素复合物1的机制靶点。NLS和NES结构域分别是核定位信号和核输出信号,NLS2的磷酸化在促进p62在细胞质和细胞核之间的转运中发挥了重要作用(Pankiv et al., 2010)。阻止p62转运到细胞质会导致泛素化蛋白聚集物在细胞核内积聚,促进p62的核易位。研究表明,p62定位于细胞核中泛素化蛋白聚集物上,提示p62可能被核泛素化蛋白聚集物降解(Wang et al., 2019; Kleine et al., 2012)。PEST结构域包括脯氨酸(P)、谷氨酸(E)、丝氨酸(S)和苏氨酸(T),也称为PEST序列。在真核细胞中,PEST序列被认为是直接与泛素蛋白酶体系统(UPS)相互作用的靶点,并且PEST的磷酸化与蛋白质的快速降解和更新有关。但是,PEST序列不是p62的特征域;在一些代谢酶、转录因子和细胞周期调节蛋白中也发现了它(Schnupf et al., 2006)。LIR结构域与LC3结合,在自噬体的形成和降解中发挥关键作用(Islam et al., 2018)。DPSTGE基序(氨基酸位置349-354)(Deng et al., 2019; Sánchez-Martín et al., 2019)p62的KIR结构域与Nrf2的Neh2结构域的基序ETGE相似,负责p62和Keap1在精氨酸380、415和483位的直接相互作用(Lau et al, 2010)。因此Nrf2通路被激活。p62的基本结构如图4所示。p62是多种细胞信号通路中的枢纽蛋白,包括Nrf2、自噬、NF-kB和Caspase 8等通路。本文主要介绍p62在Keap1-Nrf2通路和自噬通路中的作用。
4.2 Nrf2通路与自噬之间的串扰
据我们所知,p62是一种自噬适配器蛋白,p62的LIR结构域在细胞质中与LC3结合,对自噬的调节和自噬体膜的延伸具有积极作用(Lamark et al., 2017; Jiang and Mizushima, 2015)。最近的研究发现,p62参与调控Nrf2通路,使Nrf2易位到细胞核,激活下游的抗氧化基因(Bartolini et al., 2018)。p62调节氧化应激反应中的Nrf2主要有3种机制。(1)磷酸化p62的KIR结构域可能与Keap1的DGR结构域相识别,形成p62-Keap1复合物,并被UPS途径清除;(2) p62通过组装p62Keap1复合物和LC3形成LC3-p62-Keap1复合物来控制Keap1的周转率,LC3-p62-Keap1复合物被选择性自噬消除(Liao et al., 2019); (3)游离p62直接进入细胞核调控ARE,启动下游基因的表达。当细胞受到ROS或亲电体刺激时,p62与Keap1 DGR结构域的结合亲和力可通过磷酸化p62 KIR区域内的S351/349显著提高(Schnupf et al., 2006)。此外,p62过表达可通过与Ub链48K结合,通过UPS清除p62-Keap1复合物,显著降低Keap1的半衰期(Copple et al., 2010)。此外,Nrf2的DLG基序与Keap1的DGR结构域相互作用的解离有助于LC3-p62-Keap1聚集体的形成。这些聚集物被63- k连接的多聚素选择性修饰,并被包裹在自噬体中,通过自噬途径促进Keap1的降解(Dikic, 2017)。此外,Sestrin-2与ULK1相互作用,诱导p62在S409位点的UBA结构域磷酸化(Ro et al., 2014; Rhee and Bae, 2015);并显著促进LC3-p62-Keap1聚集物的自噬降解,从而上调Nrf2信号通路(Yang et al., 2020)。一些研究表明,氧化应激介导的p62蛋白水平的升高会产生p62- keap1 -Nrf2正反馈回路,导致Nrf2的持续激活(Polonen et al., 2019)。
有趣的是,Nrf2也能刺激自噬。当大量的Nrf2把进入细胞核,它是通过小加结合蛋白质的转录上调自噬相关基因,包括Atg5 p62, Map1lc3b,启动者中包括的核苷酸序列,从而上调 Atg 5,p62, LC3B蛋白的表达 (Frias et al., 2020;Ho and Gorski, 2019)。最近的研究表明Nrf2与ARE结合后还可以诱导蛋白酶体、自噬相关基因、Sestrin2和p62的表达(Dikic, 2017)。此外,Sestrin2可以通过抑制mTORC1的表达来激活自噬(Pasha et al., 2017)。因此Nrf2可直接或间接触发选择性自噬。综上所述,Nrf2通路通过p62-Keap1-Nrf2正反馈回路与自噬之间存在相互调节关系。
5. p62-Keap1-Nrf2 轴在AD中的作用
神经退行性疾病也称为蛋白质聚集性疾病。典型的神经退行性疾病有阿尔茨海默病(AD)、帕金森病(PD)、亨廷顿病(HD)、肌萎缩性侧索硬化症(ALS)和多发性硬化症(MS)。神经退行性疾病的发病机制是慢性,渐进,和不可逆转的中枢神经系统(CNS)或周围神经系统的结构和功能的变性(Poga?nik et al., 2020)。越来越多的证据表明,大量的ROS和亲电体是导致神经退行性疾病发生发展的主要因素。目前,治疗神经退行性疾病的一个有前景的策略是增加抗氧化基因的表达,包括解毒蛋白和抗氧化酶(Singh and Devasahayam, 2020)。研究表明Nrf2通路在调节神经退行性疾病中具有重要意义。已经证实,Nrf2通路的激活可以诱导细胞抗氧化保护,减少神经损伤,延缓神经退行性疾病的病情进展(Leung et al., 2019)。自噬通过参与衰老细胞器的消除和错误折叠蛋白的降解,在维持神经细胞在中枢神经系统内的稳态中发挥重要作用(Corti et al., 2020)。神经变性疾病与Nrf2通路抑制和自噬功能障碍有关,导致ROS积累、细胞器衰老和错误折叠蛋白(Darios and Stevanin, 2020; Kim S., Indu et al., 2020)。本文主要阐述了Nrf2通路和自噬在AD中的作用。
5.1 Nrf2通路对AD的影响
AD的发病机制以大脑皮层和海马的神经元和突触结构受损为特征。AD的主要组织学特征是细胞外低聚淀粉样蛋白肽(Aβ)的积累和沉积以及tau蛋白的过度磷酸化,它们与神经纤维缠结(NFT)的形成并行。这些异常蛋白的增加导致神经退行性变、神经元丢失,最终导致AD (Fão et al., 2019; Gulisano et al., 2018)。一些研究表明,活性氧(ROS)的产生和神经元损伤与活性氧的积累有关。Nrf2的表达随着年龄的增长而显著下降,细胞质中的ROS不能立即清除,这是AD的主要原因之一。这一观察结果提示Nrf2可能是AD的潜在治疗靶点(Kahroba and Davatgaran-Taghipour, 2020)。此外,在AD患者大脑中,Nrf2调控的抗氧化酶SOD1和GSH-Px活性降低(Omar et al., 1999)。当SOD和谷胱甘肽过氧化物酶(GSHJournal Px) 在AD模型显著增加,可以降低氧化应激损伤,抑制过度磷酸化tau,减少炎症推迟AD发展,增加AD小鼠的学习和记忆能力(Ji et al., 2020; Gu et al., 2020)。同样,Nrf2敲除显著影响AD小鼠的空间学习和记忆,并表现出与衰老AD患者相同的症状。Nrf2相关通路激活剂对AD有较好的治疗效果(Rojo et al., 2017; Wei et al., 2020),这表明Nrf2通路在治疗AD中是有益的。
5.2自噬对AD的影响
在AD患者中,选择性自噬与Aβ和过度磷酸化tau蛋白的消除有关。自噬抑制会降低记忆和认知能力,因此自噬受阻是AD的另一个主要原因(Li et al., 2020)。已证实用相应的激活物触发自噬有助于减少AD病理进展(Vegh et al., 2019; Song et al., 2020)。此外,Nrf2通路和选择性自噬参与了一个正反馈回路。有证据表明Nrf2的激活与神经退行性疾病中多聚素化蛋白p62的调控有关。自噬适配器p62直接与Keap1相互作用,从而释放Nrf2并将其导向细胞核。然后Nrf2与ARE结合,增加编码抗氧化酶基因的转录。有趣的是,Nrf2也会上调p62基因转录,从而产生p62- keap1 -Nrf2正反馈轴,导致Nrf2的持续激活(Pajares et al., 2016)。越来越多的研究表明,自噬的激活和p62-Keap1-Nrf2正反馈回路是改善神经退行性疾病发展的保护机制(Buratta et al., 2020; Lattante et al., 2015)。因此,自噬与Nrf2通路之间的串扰可能是治疗神经退行性疾病的一个新的靶点。其调节机制如图5所示。
综上所述,由于衰老可降低Nrf2的活性或抑制自噬,如细胞外低聚体Aβ、细胞内NFT和过度磷酸化的tau蛋白不能及时清除,会导致大量ROS的产生,进而造成海马和突触结构损伤;这是AD的主要发病机制。p62-Keap1-Nrf2正反馈回路是Nrf2通路与自噬之间的桥梁,在AD的发生发展中发挥重要作用。天然活性物质可以长期服用,因为它们产生的副作用更少。为了探讨利用天然产物靶向Nrf2通路、自噬和p62-Keap1-Nrf2反馈回路治疗AD的研究现状,我们检索了2018年1月1日至2020年4月20日的相关英汉文献。英语文献在PubMed网站(https://pubmed.ncbi.nlm.nih.gov/)进行搜索。关键词“(阿尔茨海默病)和(p62)”共61篇,关键词“(阿尔茨海默病)和(Keap1 Nrf2)”共20篇,关键词“(阿尔茨海默病)和(p62)和(Nrf2)”共5篇。中文文献通过中国知网(CNKI)网站(https://www.cnki.net/)、万方数据网站(http://www.wanfangdata.com.cn/index.html)和VIP网站(http://qikan.cqvip.com/)的高级搜索进行检索。在主题选项下,关键词“(阿尔茨海默病)和(p62)”获得17篇期刊文章,关键词“(阿尔茨海默病)和(Nrf2)”获得10篇期刊文章,关键词“((阿尔茨海默病)和(p62)和(Nrf2)”未获得任何文章。每篇文章后手动检查排除不符合需求的研究,我们发现总共有19篇文章关于天然化合物或中药配方能够激活Nrf2途径治疗AD,13个文章通过p62调节自噬干预AD,而且只有1篇关于调节Nrf2通路和自噬干预AD。具体总结见表1-2。综上所述,通过靶向p62-Keap1-Nrf2反馈回路治疗AD的策略研究,有望为未来新药的开发提供新的前景。
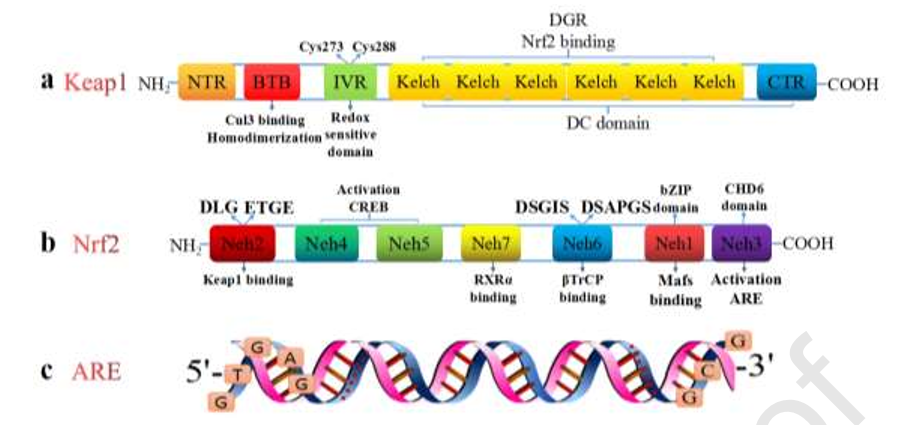
图1为Keap1 Nrf2的基本结构。a. Keap1含有624个氨基酸,由5个功能区组成。BTB、IVR和Keleh/DGR域是Keap1的重要功能域。IVR结构域含有大量半胱氨酸残基,其中Cys273和Cys288是重要位点。该区域对氧化应激敏感,也与Nrf2的稳定性有关。BTB结构域包括一个重要的应激敏感位点(Cys151),可以形成一个同源二聚体并与Cul3结合形成E3 Ub连接酶复合物。Keap1同型二聚体的DGR结构域与Nrf2的Neh2结构域的DLG和ETGE基序相互作用,对Nrf2的活性产生负调控作用。DGR和CTR统称为DC区域,是Keap1与细胞质肌动蛋白的结合区域。b. Nrf2含有624个氨基酸,可分为7个Neh结构域。Neh1结构域可以识别sMaf,并有助于Nrf2与DNA中的ARE结合。Neh2结构域包含促进Nrf2与Keap1结合的ETGE和DLG基序,介导Nrf2的降解,负向调节Nrf2的转录活性。Nrf2 c末端的Neh3区域是一个重要的功能区域,可以结合CHD6并激活ARE来调节相关基因的转录。Neh4和Neh5结构域可与CREB结合,协助Nrf2转位进入细胞核。Neh6区域的DSGIS和DSAPGS基序可以被β-TrCP识别,这对Nrf2转录有负面影响。Neh7域可以实现对RXR的识别。c. ARE位于启动子抗氧化和自噬相关基因的上游5'端区域,包括一个特定的DNA启动子结合序列。不同细胞的ARE略有不同,但共同的核苷酸序列为5'- (G/A) TGA (G/C) XXX GC (G/A) -3' (x代表任何核苷酸)。
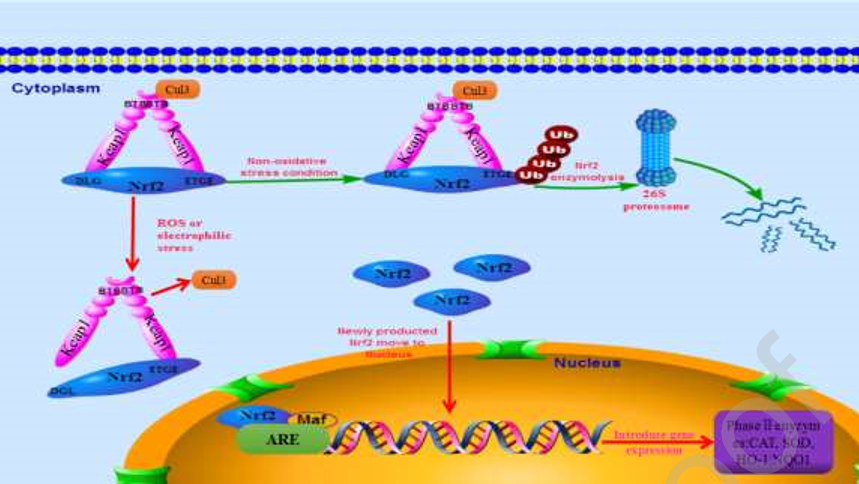
图2 Keap1-Nrf2-ARE通路的调控机制。在生理条件下,Keap1的BTB结构域与Cul3 E3 Ub连接酶复合物结合,两个Keap1分子通过BTB区域形成一个同源二聚体。Keap1-Keap1复合物与Nrf2在Keap1的DGR域以及Nrf2的ETGE和DLG基序以1:1的比例结合。Nrf2 Neh6区域的DSGIS和DSAPGS基序与E3 Ub连接酶结合,然后将Ub-蛋白酶体上的Ub转移到Nrf2的ETGE和DLG基序之间的赖氨酸残基上,诱导泛素介导的Nrf2降解。在氧化应激条件下,IVR区域的空间构象发生改变。Nrf2的弱、低亲和基序DLG与Keap1分离。Keap1 BTB结构域的Cys151被修饰,导致Keap1和Cul3的解离。游离的Nrf2易位到细胞核并与小Maf蛋白结合。Nrf2-Maf与ARE结合,增加抗氧化蛋白和II期解毒酶的转录激活。
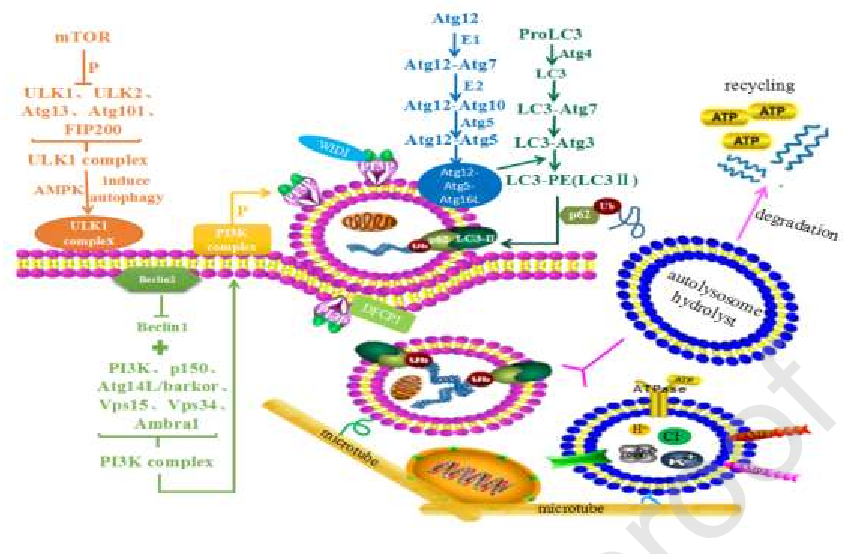
图3自噬过程。自噬可分为四个主要阶段:自噬的启动和诱导、自噬体的扩展和形成、自噬体的融合和降解、降解产物的循环。ULK1复合物、AMPK和PI3K复合物诱导自噬的启动。延长相需要Atg12-Atg5-Atg16L和Atg8/LC3偶联体系。溶酶体向MTOC移动,加速了自噬体和溶酶体相互融合的效率。包括脂肪酸和氨基酸在内的水解产物参与了细胞内的循环以提供能量。
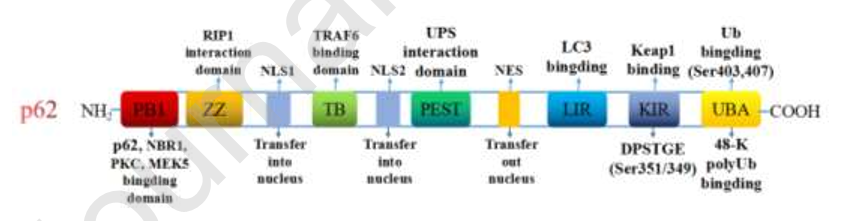
图4 p62的基本结构。p62通过PB1结构域的自寡聚形成聚集体,该结构域还与PKC、MEK5和NBR1相互作用形成异源二聚体。UBA结构域对48-K聚泛素具有高亲和力,S403和S407的磷酸化对自噬小体中泛素化底物的隔离至关重要。ZZ域与RIP1调节肿瘤坏死通路和NF-κB信号。TB结构域与TRAF6结合,催化多聚泛素链的形成。NLS域和NES域分别是核定位信号和核输出信号。PEST区域直接与UPS相互作用,而PEST的磷酸化与蛋白质的快速降解和更新有关。LIR和KIR结构域分别与LC3和Keap1结合并参与自噬。
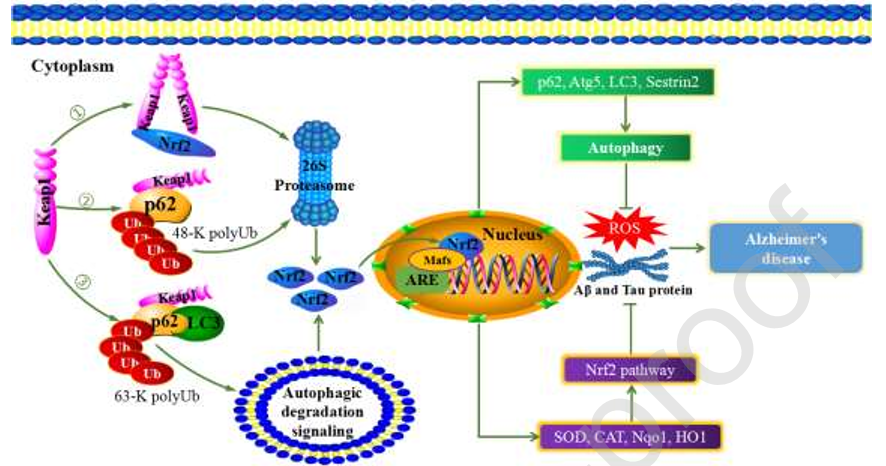
图5神经退行性疾病中Nrf2通路与自噬之间的串扰。Nrf2在氧化应激反应中主要有3种调控机制。(1) Nrf2的弱、低亲和力DLG基序与Keap1分离。Keap1的BTB结构域被修改,导致Keap1被UPS途径清除。(2)磷酸化p62的KIR结构域识别Keap1的DGR结构域,形成p62-Keap1复合物,并被UPS途径清除。(3) p62通过将p62-Keap1和LC3组装成LC3-p62-Keap1复合物来控制Keap1的周转,并通过选择性自噬降解。通过这些途径释放的Nrf2允许其转位到细胞核,并通过Maf蛋白与ARE结合。一方面,增加SOD、CAT、Nqo1和HO1的表达可以减少ROS的积累。另一方面,促进p62、Atg5、LC3和Sestrin2的转录激活自噬并降解蛋白聚集物。Nrf2通路的激活和选择性自噬可以缓解AD的发展。
翻译:
阿尔茨海默病治疗的新靶点:Nrf2通路与自噬之间的串扰
亮点:
• p62连接Nrf2通路和自噬以维持体内稳态。
• Nrf2的激活和自噬有利于AD治疗。
• p62-Keap1-Nrf2轴是治疗AD的新型潜在机制。
• 激活p62-Keap1-Nrf2轴的天然化合物是治疗AD的有效药物。
摘要:
在哺乳动物中,Keap1-Nrf2-ARE途径(以下简称“ Nrf2途径”)和自噬是主要的细胞内防御系统,可对抗氧化损伤并维持体内平衡。p62 / SQSTM1是一种泛素结合的自噬受体蛋白,它连接Nrf2途径和自噬。p62的磷酸化显著增强了其对Keap1的亲和力,从而诱导Keap1释放Nrf2,而p62-Keap1异二聚体募集了LC3并介导了Keap1在选择性自噬途径中的永久降解。最终,Nrf2积累在细胞质中,然后转移到细胞核中以激活下游编码抗氧化酶基因的转录,从而保护细胞免受氧化损伤。由于Nrf2也上调了p62基因的表达,因此创建了p62-Keap1-Nrf2正反馈环,该环进一步增强了对细胞的保护作用。研究表明,p62激活的非经典Nrf2途径是神经变性疾病的重要标志。p62-Keap1-Nrf2正反馈回路和Nrf2通路参与消除AD诱导的ROS和蛋白质聚集。因此,维持p62Keap1-Nrf2正反馈环路(是Nrf2途径与自噬之间的桥梁)的体内平衡可能是治疗AD的潜在目标。
关键词:Nrf2通路、自噬、氧化应激、p62-Keap1-Nrf2正反馈回路、阿尔茨海默病
1. 前言
活性氧(ROS)是正常有氧代谢的副产物,在多种危险因素中产生,如环境应激、衰老和疾病。高水平的ROS破坏DNA结构和膜分布,改变蛋白质结构和功能,导致细胞损伤和死亡。氧化应激反应清除体内过多的ROS,维持体内氧化还原稳态(Yi et al., 2020; Xia et al., 2020)。核因子、红细胞来源因子2相关因子(Nrf2)是抗氧化防御的关键转录因子,与Kelch like ech相关蛋白1 (Keap1)相互作用。在氧化应激条件下,由于过多的活性氧产量,Nrf2 Keap1分离,进入细胞核,它结合抗氧化反应的元素(ARE)触发下游抗氧化酶的表达,如血红素加氧酶1 (HO1)、超氧化物歧化酶(SOD)、过氧化氢酶(CAT)和NAD (P) H醌脱氢酶1 (NQO1)。
自噬在真核细胞中广泛存在,是一种自我修复的生物过程,参与细胞生长、代谢和抗氧化应激。自噬负责清除长寿命或错误折叠的蛋白质、脂滴和受损的细胞器,以保持细胞内稳态(Liu et al., 2020)。自噬是一种溶酶体依赖的自我降解途径。根据底物运输到溶酶体进行降解的方式,自噬途径通常分为三种不同类型,这三种类型分别是微自噬、伴侣介导的自噬(CMA,只发生在哺乳动物)和巨自噬(Suzuki et al., 2017)。在微自噬过程中,底物被运输到溶酶体中,并被小的溶酶体膜的内陷所隔离,这些内陷被挤压到内腔中(Li and Hochstrasser, 2020)。在CMA中,五肽基序(KFERQ)暴露的错误折叠蛋白被热休克70 kDa蛋白8 (HSPA8/HSC70)识别,进而与溶酶体相关膜蛋白2A (LAMP2A)结合。这些蛋白在溶酶体腔内展开和降解(Dou et al., 2020)。自噬最典型的形式是巨自噬(以下简称自噬)。自噬的特点是胞质成分包裹在称为自噬小体的双层膜结构中。然后,自噬体与溶酶体膜融合并被水解酶降解。在营养缺乏的情况下,该途径被激活以提供氨基酸和ATP (Yang et al., 2019)。
p62转录被氧化应激、代谢紊乱、疾病和自噬受阻激活,导致细胞质中大量p62聚集。p62的聚集物通过与Nrf2抑制蛋白Keap1直接相互作用,维持Nrf2的慢性激活,并且p62介导Keap1在选择性自噬途径中的永久降解(Sanchez-Martin and Komatsu, 2018)。因此,Nrf2信号通路被激活,导致编码抗氧化酶的基因转录上调(Deng et al., 2020)。此外,核内Nrf2促进p62基因的过表达,形成p62- keap1 -Nrf2正反馈轴,导致Nrf2的持续激活。与经典途径相比,p62介导的选择性自噬调节Nrf2信号通路属于非经典机制。越来越多的研究表明,p62-Keap1-Nrf2通路的失调与神经退行性疾病的治疗有关(Shah et al., 2018)。因此,本综述将阐述Nrf2通路、自噬及其串扰在AD治疗中的作用。
2. Nrf2 通路
2.1 Keap1的基本结构
Keap1的分子量为66 kDa,由624个氨基酸组成。它是Cullin3 (Cul3)-E3泛素连接酶复合物的底物蛋白,是一种负向调节Nrf2活性的胞质蛋白(Davies et al., 2016)。从结构上看,Keap1由5个功能区组成。第一个区域是N端区域(NTR)。第二个区域是间质区(IVR),其中含有大量的半胱氨酸残基。第三个区域是brac -a-brac (BTB)区域,包括broad complex、tramtrack和brac,其中包括一个重要的应力敏感位点半胱氨酸151 (Cys151)。后两个区域是双甘氨酸重复或Keleh重复(DGR)区域和C末端区域(CTR) (Dinkovakostova et al., 2017; Deshmukh et al., 2017)。在这些区域中,BTB、IVR和Keleh/DGR域是Keap1的重要功能域。BTB结构域是蛋白质-蛋白质相互作用中一个进化保守的序列。这个结构域可以与另一个Keap1分子的BTB区域结合,形成一个同分二聚体,然后与Nrf2结合。该区域还可以与Cul3结合形成E3 Ub连接酶复合物,导致泛素介导的Nrf2降解(Kansanen et al., 2012; Tao et al., 2017)。在BTB结构域,当半胱氨酸残基的结构发生改变时,Nrf2蛋白的泛素化和降解将受到抑制。这一过程诱导Nrf2与Keap1分离并与ARE结合以激活编码抗氧化酶的基因转录。IVR区域富含半胱氨酸残基,对氧化应激敏感,也与Nrf2的稳定性有关。在正常生理条件下,IVR区域参与泛素介导的Nrf2降解。值得一提的是,Cys273和Cys288在抑制Nrf2活性方面发挥了重要作用。当机体被亲电体和ROS激活时,IVR结构域发生转变,导致Nrf2和Keap1相互分离(Magesh et al., 2012)。DGR结构域(包括6个Kelch重复)和CTR区域统称为DC结构域,是Keap1与细胞质肌动蛋白的结合位点。Keap1同型二聚体中的DGR结构域与Nrf2中Neh2结构域的DLG和ETGE氨基酸基序相互作用,对Nrf2活性产生负调控作用。在外源性或内源性刺激下,通过半胱氨酸失活诱导Keap1 DGR区域构象变化,诱导Keap1-Nrf2蛋白复合物解离,释放Nrf2进入细胞核。Keap1的基本结构如图1a所示。
2.2 Nrf2的基本结构
Nrf2分子量为100kda,由624个氨基酸组成。Nrf2属于碱性亮氨酸拉链(basic leucine zipper, bZIP)转录因子家族,其成员保持cap-n-collar (CNC)结构,而人类Nfe2l2位于2q31上(Tavakkoli et al., 2019; Bottino-Rojas et al., 2018)。根据其生理功能,Nrf2可分为7个高度保守的ECH同源结构域(Nrf2- ECH同源),包括Neh1到Neh7 (ECH,与鸡Nrf2具有CNC同源性的红血球来源蛋白)。亮氨酸CNC-bZIP区域位于Neh1结构域,可与细胞核中的一种小的肌肉筋膜性纤维肉瘤(Maf)蛋白(即脊椎动物中的MafF、MafG或MafK)结合,并有助于Nrf2与DNA中的ARE结合。随后,抗氧化酶、Ub酶、II期解毒酶和蛋白酶体的转录上调,以抵御细胞中的氧化应激(Karan et al., 2020; Shen et al., 2019)。Neh2结构域是一个由16个氨基酸组成的短肽,其氨基酸序列在69~84之间,位于Nrf2的N端。Nrf2的Neh2结构域包含分别与Keap1具有高亲和力和低亲和力的ETGE和DLG基序;该区域介导Nrf2的降解并负调控Nrf2的转录活性(Tian et al., 2018)。Nrf2 C -末端的Neh3区域是一个可以结合CHD6并激活ARE的重要功能区域(Zhang et al., 2014)。Neh4和Neh5域可以结合激活cAMP反应元素结合蛋白(CREB)协助Nrf2核转运和以Nrf2-Maf的形式绑定到ARE (Krajka Ku?niak et al ., 2016)。Neh6结构域具有大量的丝氨酸残基(Chen et al., 2020),对Nrf2转录有负向影响,且不依赖于Keap1 (Chowdhry et al., 2013),该结构域主要参与Nrf2的降解。其调控机制是DSGIS和DSAPGS的氨基酸基序可通过β-转介含重复蛋白(β-TrCP)识别,并可诱导Skp1-Cul1-Rbx1/Roc1-β-TrCP Ub连接酶复合物调控Nrf2泛素化介导的降解(Cuadrado, 2015)。Neh7结构域识别视黄素X受体(RXR) (Li et al., 2017)。此外,Nrf2的第40个氨基酸是一个磷酸化位点,该位点的磷酸化导致Nrf2从Keap1中解离。Nrf2的基本结构如图1b所示。
2.3 ARE的基本结构
ARE位于一些细胞保护基因(抗氧化和自噬相关基因)启动子区域上游5'端,属于特定的DNA启动子联合序列(Jeddi et al., 2017)。ARE被定义为顺式作用的DNA增强因子,负责触发氧化应激反应,以诱导II期解毒酶和抗氧化酶的表达。ARE广泛表达于生物体中,其功能是减轻细胞或组织的各种损伤,维持体内平衡。虽然在不同的细胞中ARE略有不同,但共同的核苷酸序列为5'- (G/A) TGA (G/C) XXX GC (G/A) -3' (x代表任何核苷酸)。ARE的基本结构如图1c所示。
2.4 Nrf2信号通路的调控机制
在漫长的进化过程中,细胞获得了抵抗外部毒素和内部代谢毒素的能力。最重要的转录因子Nrf2调节抗氧化应激反应(Erukainure et al., 2020)[29]。Keap1-Nrf2- ARE通路是激活Nrf2的典型调控通路,Nrf2的活性主要由Keap1调控。在基础条件下,Keap1的BTB结构域与Cul3 E3 Ub连接酶复合物结合,两个Keap1分子通过BTB区域形成一个同源二聚体。Keap1-Keap1复合物通过Keap1的DGR域与Nrf2的ETGE和DLG基序以1:1的比例直接结合到Nrf2上(Raghunath et al., 2019)。Nrf2 Neh6域的DSGIS和DSAPGS氨基酸基序与E3 Ub连接酶结合,然后, Ub-蛋白酶体上得Ub转移到Nrf2ETGE、DLG基序之间的赖氨酸残基诱导泛素介导的Nrf2降解,因此保持适当的浓度Nrf2(Lee and Ryu, 2017)。
在氧化应激条件下,线粒体产生大量的高活性分子,如ROS (Chen et al., 2020);当ROS含量超过细胞清除能力时,氧化还原系统出现失衡,导致脂质、蛋白质和DNA发生氧化损伤,最终导致细胞凋亡、组织或器官损伤(Liu et al., 2017; Valavanidis et al., 2013)。因此Nrf2通路被显著激活,具体机制如下。一方面,Keap1 IVR区Cys273和Cys288的巯基被氧化形成二硫键(Magesh et al., 2012),导致空间构象的改变。然后,具有较强结合能力的高亲和力ETGE 基序仍然能够与Keap1紧密结合,而较弱、低亲和力的DLG 基序与Keap1分离。Nrf2的空间定位发生改变,不能被泛素蛋白酶体降解。另一方面,Keap1 BTB结构域的Cys151被修饰,产生空间位阻效应,导致Keap1和Cul3解离(Wang et al., 2020)。这破坏了keap1-cul3 E3 泛素连接酶的活性,并抑制了泛素介导的Nrf2降解。当Keap1-Keap1-Nrf2的数量达到饱和时,Keap1同型二聚体在细胞中不能再生,新生成的Nrf2由于自身的保护,不再与Keap1结合。Nrf2迁移到细胞核并与小Maf蛋白结合。然后,Nrf2-Maf与ARE结合,增加抗氧化蛋白和II期解毒酶的转录激活(Otsuki and Yamamoto, 2019)。当氧化还原平衡恢复后,Nrf2从细胞核转位到细胞质,并通过泛素化降解(Pandey et al., 2017)。Nrf2通路的具体调控机制如图2所示。
3. 自噬的调节机制
从机制上看,自噬主要分为四个阶段,即自噬的启动和诱导、自噬体的扩展和形成、自噬体与溶酶体融合和降解以及降解产物的循环利用(Carlsson and Simonsen, 2015)。在营养充足的条件下,哺乳动物雷帕霉素靶蛋白(mTOR)是自噬的重要调节因子,其作用是抑制自噬。mTOR存在于细胞内的两个复合物中;这些复合物是mTOR复合物1 (mTORC1)和mTOR复合物2 (mTORC2)。mTORC1复合物由mTOR、raptor和富含脯氨酸的Akt底物(PRAS40)组成,raptor是mTORC1激活的重要因素。此外,mTOR、rictor、哺乳动物应激激活的MAP激酶相互作用蛋白(mSIN1)以及与rictor 1和2观察到的蛋白形成mTORC2复合物。unc -51样激酶1 (ULK1)是自噬核心机制中最上游的激酶,ULK1的磷酸化是自噬激活的重要决定因素。AMP活化蛋白激酶(AMPK)和mTORC1可以直接调节ULK1 (Atg1 homologs)的磷酸化,这些蛋白是自噬的重要调节蛋白。AMPK正调控自噬,mTORC1通过与ULK1结合抑制ULK1复合物的磷酸化,从而抑制自噬(Noda, 2017)。
3.1自噬的启动和诱导
正常情况下,自噬被mTORC1抑制,Beclin1与Beclin2在内质网(ER)上形成复合物。在缺乏营养或生长因子的条件下,自噬被认为是非选择性的,并集中于增加任何胞质蛋白和其他大分子的降解,以提供必需的营养物质。在应激条件下,自噬被认为是选择性的,有目的地识别底物并将其紧密地结合到新出现的自噬体膜上。相关机制如下。mTORC1失活后,由ULK1、ULK2、Atg13、Atg101和局灶粘附激酶家族相互作用蛋白(FIP200) (Neufeld, 2020)组成的ULK1复合物被AMPK激活后转运到ER (Chen et al, 2020)。磷酸化的ULK1复合物调控JNK1酶诱导Beclin2的磷酸化,从而改变Beclin2的空间构像,将Beclin1释放到细胞质中(Xue et al., 2015)。Beclin1对应激高度敏感(Kiruthiga et al., 2020),并与III类磷脂酰肌醇3激酶(IIIPI3K/PIK3C3;酵母中的Vps34), Atg14L(也称为BARKOR;酵母中的Atg14), p150(又称PIK3R4;酵母菌Vps15)和AMBRA1。PI3K (PI3P)的磷酸化参与了自噬的启动(Nascimbeni et al., 2017)。部分PI3P复合物征募双FYVE-containing protein 1 (DECP1),被ER包裹以增强膜的弯曲度,并形成一个收集自噬相关蛋白的平台。PI3P与WD重复域磷酸肌醇肽相互作用的另一部分(WIPI, Atg18同源物)在胞浆-液泡(Cvt)通路中发挥重要作用。这个阶段是自噬前体膜的形成(Cao et al., 2019;Nakatogawa, 2020)。
3.2自噬体的延伸和形成
自噬前体的双层膜被不断拉长以形成自噬体。在延长阶段需要两个Atg接合系统被ub -蛋白酶体修饰。一个系统是Atg12-Atg5-Atg16L共轭系统,锚定自噬体的外膜,另一个系统是Atg8/LC3共轭系统,同时连接内膜和外膜。Atg12-Atg5-Atg16L复合物是自噬体形成所必需的。自噬激活后,ub样蛋白Atg12被e1样ub激活酶和e2样ub结合酶诱导,通过Atg7和Atg10作用与Atg5结合。Atg16的低聚物与Atg12-Atg5的共轭物结合,形成位于外膜的Atg12-Atg5- atg16l复合物(Liet al., 2020)。在非自噬条件下,LC3(在酵母中也被称为Atg8同源蛋白)以proLC3的非活性形式存在。自噬激活后,LC3通过Atg4、Atg7、Atg3的顺序作用转化为LC3I。然后Atg12-Atg5-Atg16L复合物介导LC3I与磷脂酰乙醇胺(PE)结合,形成具有膜结合能力的活性蛋白LC3II。LC3II与p62的LIR结构域相互作用,影响扩张吞噬团的曲率和自噬前体的形成。自噬前体膜通过诱导LC3II不断延伸形成囊状自噬体,嵌入双层膜表面(Noda and Inagaki, 2015)。
3.3自噬体与溶酶体的融合和产物降解
自噬体与溶酶体的融合是通过两种机制:一个机制是直接的自噬体与溶酶体融合形成自吞噬泡,和其他机制的结合与成熟的核内体自噬体形成自噬核内体,后来搬到溶酶体与溶酶体融合。细胞核周围的微管组织中心(MTOC)含有大量的溶酶体。在压力作用下,膜附LC3-II蛋白在肌动蛋白复合物的作用下移动到MTOC附近。同时,溶酶体向MTOC移动,加速了相互融合的效率。在自噬溶酶体形成初期,ATP为溶酶体膜上的质子泵提供能量,增加溶酶体主动摄取H+。阳离子和阴离子泵可以维持溶酶体中的离子电流平衡(如Ca2+、K+和Cl-)。当内溶酶体的pH值在4.5 ~ 5.0时,水解酶被激活,困在自噬体内的LC3II蛋白与底物一起最终被降解。自噬体外膜上LC3-II被Atg4降解回收。基于多糖的膜蛋白(如LAMP1和LAMP2)嵌入溶酶体膜表面,可以防止溶酶体自身降解。自噬溶酶体融合后,水解酶降解底物,包括脂肪酸和氨基酸在内的水解物参与细胞内循环,提供能量(Zhao et al., 2020)。具体流程如图3所示。
4. 细胞中的p62-Keap1-Nrf2轴
4.1 p62的基本结构
p62,又称SQSTM1,是一种与ub结合的自噬受体,分子量为62 kDa。p62主要位于细胞质中,可在细胞质与细胞核之间转运。p62含有440个氨基酸,由9功能域:phox bem1域(PB1), ZZ-type锌指域(ZZ) TRAF6绑定域(TB),核本地化信号(NLS),核出口信号(NES), PEST域,LC3交互领域(LIR), Keap1相互作用域(KIR)和C末端 Ub相关(UBA)域(Lin et al., 2013)。p62通过PB1结构域的selfoligomerization形成聚合体,该结构域还与PKC、胞外信号调节激酶5 (MEK5)和邻近的BRCA1基因1蛋白(NBR1)相互作用,形成异源二聚体。c端UBA结构域,对Ub具有高亲和力的高度保守序列(Katsuragi et al., 2015; Denk et al., 2019),对于隔离转运到自噬体中的泛素化底物至关重要。此外,UBA结构域的S403和S407可被靶向形成Ub链48K的蛋白聚合物(Zhang et al., 2019),并被由两个亚基组成的26S蛋白酶体识别,这两个亚基是一个20S蛋白酶体和一个19S调节粒子。19S粒子识别泛素化的p62蛋白,并通过狭窄的孔将其转移至20S蛋白酶体,20S蛋白酶体将底物消化为包含2-24个氨基酸的肽,并为细胞再利用提供必要的材料(Jakobi et al., 2020; Cohen-Kaplan et al., 2017)。ZZ域与受体相互作用蛋白1 (RIP1)调节肿瘤坏死通路和NF-κB信号(Yan et al ., 2017)。TB结构域结合TRAF6催化赖氨酸(K)残基位点(63位)多聚泛素链的形成(Kim et al., 2019)。ZZ和TB区域之间的未知区域与raptor相互作用,激活雷帕霉素复合物1的机制靶点。NLS和NES结构域分别是核定位信号和核输出信号,NLS2的磷酸化在促进p62在细胞质和细胞核之间的转运中发挥了重要作用(Pankiv et al., 2010)。阻止p62转运到细胞质会导致泛素化蛋白聚集物在细胞核内积聚,促进p62的核易位。研究表明,p62定位于细胞核中泛素化蛋白聚集物上,提示p62可能被核泛素化蛋白聚集物降解(Wang et al., 2019; Kleine et al., 2012)。PEST结构域包括脯氨酸(P)、谷氨酸(E)、丝氨酸(S)和苏氨酸(T),也称为PEST序列。在真核细胞中,PEST序列被认为是直接与泛素蛋白酶体系统(UPS)相互作用的靶点,并且PEST的磷酸化与蛋白质的快速降解和更新有关。但是,PEST序列不是p62的特征域;在一些代谢酶、转录因子和细胞周期调节蛋白中也发现了它(Schnupf et al., 2006)。LIR结构域与LC3结合,在自噬体的形成和降解中发挥关键作用(Islam et al., 2018)。DPSTGE基序(氨基酸位置349-354)(Deng et al., 2019; Sánchez-Martín et al., 2019)p62的KIR结构域与Nrf2的Neh2结构域的基序ETGE相似,负责p62和Keap1在精氨酸380、415和483位的直接相互作用(Lau et al, 2010)。因此Nrf2通路被激活。p62的基本结构如图4所示。p62是多种细胞信号通路中的枢纽蛋白,包括Nrf2、自噬、NF-kB和Caspase 8等通路。本文主要介绍p62在Keap1-Nrf2通路和自噬通路中的作用。
4.2 Nrf2通路与自噬之间的串扰
据我们所知,p62是一种自噬适配器蛋白,p62的LIR结构域在细胞质中与LC3结合,对自噬的调节和自噬体膜的延伸具有积极作用(Lamark et al., 2017; Jiang and Mizushima, 2015)。最近的研究发现,p62参与调控Nrf2通路,使Nrf2易位到细胞核,激活下游的抗氧化基因(Bartolini et al., 2018)。p62调节氧化应激反应中的Nrf2主要有3种机制。(1)磷酸化p62的KIR结构域可能与Keap1的DGR结构域相识别,形成p62-Keap1复合物,并被UPS途径清除;(2) p62通过组装p62Keap1复合物和LC3形成LC3-p62-Keap1复合物来控制Keap1的周转率,LC3-p62-Keap1复合物被选择性自噬消除(Liao et al., 2019); (3)游离p62直接进入细胞核调控ARE,启动下游基因的表达。当细胞受到ROS或亲电体刺激时,p62与Keap1 DGR结构域的结合亲和力可通过磷酸化p62 KIR区域内的S351/349显著提高(Schnupf et al., 2006)。此外,p62过表达可通过与Ub链48K结合,通过UPS清除p62-Keap1复合物,显著降低Keap1的半衰期(Copple et al., 2010)。此外,Nrf2的DLG基序与Keap1的DGR结构域相互作用的解离有助于LC3-p62-Keap1聚集体的形成。这些聚集物被63- k连接的多聚素选择性修饰,并被包裹在自噬体中,通过自噬途径促进Keap1的降解(Dikic, 2017)。此外,Sestrin-2与ULK1相互作用,诱导p62在S409位点的UBA结构域磷酸化(Ro et al., 2014; Rhee and Bae, 2015);并显著促进LC3-p62-Keap1聚集物的自噬降解,从而上调Nrf2信号通路(Yang et al., 2020)。一些研究表明,氧化应激介导的p62蛋白水平的升高会产生p62- keap1 -Nrf2正反馈回路,导致Nrf2的持续激活(Polonen et al., 2019)。
有趣的是,Nrf2也能刺激自噬。当大量的Nrf2把进入细胞核,它是通过小加结合蛋白质的转录上调自噬相关基因,包括Atg5 p62, Map1lc3b,启动者中包括的核苷酸序列,从而上调 Atg 5,p62, LC3B蛋白的表达 (Frias et al., 2020;Ho and Gorski, 2019)。最近的研究表明Nrf2与ARE结合后还可以诱导蛋白酶体、自噬相关基因、Sestrin2和p62的表达(Dikic, 2017)。此外,Sestrin2可以通过抑制mTORC1的表达来激活自噬(Pasha et al., 2017)。因此Nrf2可直接或间接触发选择性自噬。综上所述,Nrf2通路通过p62-Keap1-Nrf2正反馈回路与自噬之间存在相互调节关系。
5. p62-Keap1-Nrf2 轴在AD中的作用
神经退行性疾病也称为蛋白质聚集性疾病。典型的神经退行性疾病有阿尔茨海默病(AD)、帕金森病(PD)、亨廷顿病(HD)、肌萎缩性侧索硬化症(ALS)和多发性硬化症(MS)。神经退行性疾病的发病机制是慢性,渐进,和不可逆转的中枢神经系统(CNS)或周围神经系统的结构和功能的变性(Poga?nik et al., 2020)。越来越多的证据表明,大量的ROS和亲电体是导致神经退行性疾病发生发展的主要因素。目前,治疗神经退行性疾病的一个有前景的策略是增加抗氧化基因的表达,包括解毒蛋白和抗氧化酶(Singh and Devasahayam, 2020)。研究表明Nrf2通路在调节神经退行性疾病中具有重要意义。已经证实,Nrf2通路的激活可以诱导细胞抗氧化保护,减少神经损伤,延缓神经退行性疾病的病情进展(Leung et al., 2019)。自噬通过参与衰老细胞器的消除和错误折叠蛋白的降解,在维持神经细胞在中枢神经系统内的稳态中发挥重要作用(Corti et al., 2020)。神经变性疾病与Nrf2通路抑制和自噬功能障碍有关,导致ROS积累、细胞器衰老和错误折叠蛋白(Darios and Stevanin, 2020; Kim S., Indu et al., 2020)。本文主要阐述了Nrf2通路和自噬在AD中的作用。
5.1 Nrf2通路对AD的影响
AD的发病机制以大脑皮层和海马的神经元和突触结构受损为特征。AD的主要组织学特征是细胞外低聚淀粉样蛋白肽(Aβ)的积累和沉积以及tau蛋白的过度磷酸化,它们与神经纤维缠结(NFT)的形成并行。这些异常蛋白的增加导致神经退行性变、神经元丢失,最终导致AD (Fão et al., 2019; Gulisano et al., 2018)。一些研究表明,活性氧(ROS)的产生和神经元损伤与活性氧的积累有关。Nrf2的表达随着年龄的增长而显著下降,细胞质中的ROS不能立即清除,这是AD的主要原因之一。这一观察结果提示Nrf2可能是AD的潜在治疗靶点(Kahroba and Davatgaran-Taghipour, 2020)。此外,在AD患者大脑中,Nrf2调控的抗氧化酶SOD1和GSH-Px活性降低(Omar et al., 1999)。当SOD和谷胱甘肽过氧化物酶(GSHJournal Px) 在AD模型显著增加,可以降低氧化应激损伤,抑制过度磷酸化tau,减少炎症推迟AD发展,增加AD小鼠的学习和记忆能力(Ji et al., 2020; Gu et al., 2020)。同样,Nrf2敲除显著影响AD小鼠的空间学习和记忆,并表现出与衰老AD患者相同的症状。Nrf2相关通路激活剂对AD有较好的治疗效果(Rojo et al., 2017; Wei et al., 2020),这表明Nrf2通路在治疗AD中是有益的。
5.2自噬对AD的影响
在AD患者中,选择性自噬与Aβ和过度磷酸化tau蛋白的消除有关。自噬抑制会降低记忆和认知能力,因此自噬受阻是AD的另一个主要原因(Li et al., 2020)。已证实用相应的激活物触发自噬有助于减少AD病理进展(Vegh et al., 2019; Song et al., 2020)。此外,Nrf2通路和选择性自噬参与了一个正反馈回路。有证据表明Nrf2的激活与神经退行性疾病中多聚素化蛋白p62的调控有关。自噬适配器p62直接与Keap1相互作用,从而释放Nrf2并将其导向细胞核。然后Nrf2与ARE结合,增加编码抗氧化酶基因的转录。有趣的是,Nrf2也会上调p62基因转录,从而产生p62- keap1 -Nrf2正反馈轴,导致Nrf2的持续激活(Pajares et al., 2016)。越来越多的研究表明,自噬的激活和p62-Keap1-Nrf2正反馈回路是改善神经退行性疾病发展的保护机制(Buratta et al., 2020; Lattante et al., 2015)。因此,自噬与Nrf2通路之间的串扰可能是治疗神经退行性疾病的一个新的靶点。其调节机制如图5所示。
综上所述,由于衰老可降低Nrf2的活性或抑制自噬,如细胞外低聚体Aβ、细胞内NFT和过度磷酸化的tau蛋白不能及时清除,会导致大量ROS的产生,进而造成海马和突触结构损伤;这是AD的主要发病机制。p62-Keap1-Nrf2正反馈回路是Nrf2通路与自噬之间的桥梁,在AD的发生发展中发挥重要作用。天然活性物质可以长期服用,因为它们产生的副作用更少。为了探讨利用天然产物靶向Nrf2通路、自噬和p62-Keap1-Nrf2反馈回路治疗AD的研究现状,我们检索了2018年1月1日至2020年4月20日的相关英汉文献。英语文献在PubMed网站(https://pubmed.ncbi.nlm.nih.gov/)进行搜索。关键词“(阿尔茨海默病)和(p62)”共61篇,关键词“(阿尔茨海默病)和(Keap1 Nrf2)”共20篇,关键词“(阿尔茨海默病)和(p62)和(Nrf2)”共5篇。中文文献通过中国知网(CNKI)网站(https://www.cnki.net/)、万方数据网站(http://www.wanfangdata.com.cn/index.html)和VIP网站(http://qikan.cqvip.com/)的高级搜索进行检索。在主题选项下,关键词“(阿尔茨海默病)和(p62)”获得17篇期刊文章,关键词“(阿尔茨海默病)和(Nrf2)”获得10篇期刊文章,关键词“((阿尔茨海默病)和(p62)和(Nrf2)”未获得任何文章。每篇文章后手动检查排除不符合需求的研究,我们发现总共有19篇文章关于天然化合物或中药配方能够激活Nrf2途径治疗AD,13个文章通过p62调节自噬干预AD,而且只有1篇关于调节Nrf2通路和自噬干预AD。具体总结见表1-2。综上所述,通过靶向p62-Keap1-Nrf2反馈回路治疗AD的策略研究,有望为未来新药的开发提供新的前景。
图1为Keap1 Nrf2的基本结构。a. Keap1含有624个氨基酸,由5个功能区组成。BTB、IVR和Keleh/DGR域是Keap1的重要功能域。IVR结构域含有大量半胱氨酸残基,其中Cys273和Cys288是重要位点。该区域对氧化应激敏感,也与Nrf2的稳定性有关。BTB结构域包括一个重要的应激敏感位点(Cys151),可以形成一个同源二聚体并与Cul3结合形成E3 Ub连接酶复合物。Keap1同型二聚体的DGR结构域与Nrf2的Neh2结构域的DLG和ETGE基序相互作用,对Nrf2的活性产生负调控作用。DGR和CTR统称为DC区域,是Keap1与细胞质肌动蛋白的结合区域。b. Nrf2含有624个氨基酸,可分为7个Neh结构域。Neh1结构域可以识别sMaf,并有助于Nrf2与DNA中的ARE结合。Neh2结构域包含促进Nrf2与Keap1结合的ETGE和DLG基序,介导Nrf2的降解,负向调节Nrf2的转录活性。Nrf2 c末端的Neh3区域是一个重要的功能区域,可以结合CHD6并激活ARE来调节相关基因的转录。Neh4和Neh5结构域可与CREB结合,协助Nrf2转位进入细胞核。Neh6区域的DSGIS和DSAPGS基序可以被β-TrCP识别,这对Nrf2转录有负面影响。Neh7域可以实现对RXR的识别。c. ARE位于启动子抗氧化和自噬相关基因的上游5'端区域,包括一个特定的DNA启动子结合序列。不同细胞的ARE略有不同,但共同的核苷酸序列为5'- (G/A) TGA (G/C) XXX GC (G/A) -3' (x代表任何核苷酸)。
图2 Keap1-Nrf2-ARE通路的调控机制。在生理条件下,Keap1的BTB结构域与Cul3 E3 Ub连接酶复合物结合,两个Keap1分子通过BTB区域形成一个同源二聚体。Keap1-Keap1复合物与Nrf2在Keap1的DGR域以及Nrf2的ETGE和DLG基序以1:1的比例结合。Nrf2 Neh6区域的DSGIS和DSAPGS基序与E3 Ub连接酶结合,然后将Ub-蛋白酶体上的Ub转移到Nrf2的ETGE和DLG基序之间的赖氨酸残基上,诱导泛素介导的Nrf2降解。在氧化应激条件下,IVR区域的空间构象发生改变。Nrf2的弱、低亲和基序DLG与Keap1分离。Keap1 BTB结构域的Cys151被修饰,导致Keap1和Cul3的解离。游离的Nrf2易位到细胞核并与小Maf蛋白结合。Nrf2-Maf与ARE结合,增加抗氧化蛋白和II期解毒酶的转录激活。
图3自噬过程。自噬可分为四个主要阶段:自噬的启动和诱导、自噬体的扩展和形成、自噬体的融合和降解、降解产物的循环。ULK1复合物、AMPK和PI3K复合物诱导自噬的启动。延长相需要Atg12-Atg5-Atg16L和Atg8/LC3偶联体系。溶酶体向MTOC移动,加速了自噬体和溶酶体相互融合的效率。包括脂肪酸和氨基酸在内的水解产物参与了细胞内的循环以提供能量。
图4 p62的基本结构。p62通过PB1结构域的自寡聚形成聚集体,该结构域还与PKC、MEK5和NBR1相互作用形成异源二聚体。UBA结构域对48-K聚泛素具有高亲和力,S403和S407的磷酸化对自噬小体中泛素化底物的隔离至关重要。ZZ域与RIP1调节肿瘤坏死通路和NF-κB信号。TB结构域与TRAF6结合,催化多聚泛素链的形成。NLS域和NES域分别是核定位信号和核输出信号。PEST区域直接与UPS相互作用,而PEST的磷酸化与蛋白质的快速降解和更新有关。LIR和KIR结构域分别与LC3和Keap1结合并参与自噬。
图5神经退行性疾病中Nrf2通路与自噬之间的串扰。Nrf2在氧化应激反应中主要有3种调控机制。(1) Nrf2的弱、低亲和力DLG基序与Keap1分离。Keap1的BTB结构域被修改,导致Keap1被UPS途径清除。(2)磷酸化p62的KIR结构域识别Keap1的DGR结构域,形成p62-Keap1复合物,并被UPS途径清除。(3) p62通过将p62-Keap1和LC3组装成LC3-p62-Keap1复合物来控制Keap1的周转,并通过选择性自噬降解。通过这些途径释放的Nrf2允许其转位到细胞核,并通过Maf蛋白与ARE结合。一方面,增加SOD、CAT、Nqo1和HO1的表达可以减少ROS的积累。另一方面,促进p62、Atg5、LC3和Sestrin2的转录激活自噬并降解蛋白聚集物。Nrf2通路的激活和选择性自噬可以缓解AD的发展。
本文由福山生物整理翻译,转载请注明出处。
下一篇:没有了!