









-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
AMPK在阿尔茨海默病中的作用机制
发表于:2023-02-24 作者:超级管理员 来源:本站 点击量:15612
阿尔茨海默病(Alzheimer’sdisease, AD)是一个与年龄密切相关的进行性神经变性疾病,是痴呆的最常见的临床表现形式。据估计,2010年在世界各地有3.56亿AD患者,2030年这将增加至6.57亿,而到2050年可达11.54亿。与日俱增的AD患者必将会给各国政府带来严重的社会问题和经济问题,对于AD的防治也备受世界各国重视。
目前有关AD的研究浪潮也是此起披伏,硕果累累。多种因素参与了AD的发病机制,不管原因如何,都会归结为AD的经典病理特征的形成:细胞外老年斑(SenilePlaques, SP)和细胞内神经纤维原缠结(NeurofibrillaryTangles, NFTs)。AD进展取决于SP和NFTs积聚和由此而引发的神经变性性病变过程。Aβ产生和沉积是一个关键的特征,也是SP形成的根本因素。NFTs是AD神经细胞内发现的不溶性扭曲纤维,主要由tau蛋白形成,是微管结构的一部分。微管有助于营养物和其他重要的物质在神经细胞内输送,在AD中tau蛋白异常,微管结构解聚。尽管已有很多研究成果运用于临床防治AD中,但还是没有治愈AD或阻止其进展的有效方法。
腺苷酸活化蛋白激酶( AMP-activated protein kinase, AMPK ) 是丝/苏氨酸蛋白激酶,一种重要的蛋白激酶,主要协调代谢和能量的需要。短期效应能调节能量代谢,长期效应能调节基因转录[1]。大量研究表明AMPK作为哺乳动物细胞代谢传感器参与了神经变性性变的调控[2]。因此,AMPK或许是神经变性疾病一种新的共同决定因素,调控AMPK活性有可能成为研发治疗AD药物的新途径。
AMPK结构和功能
AMP作为能量传感器和调节器,在调节细胞能量稳态的起着关键作用。AMPK是一个异源三聚体蛋白,由α(63kD)、β(30kD)和γ (37-63kD) 3个亚单位组成。其中,α亚单位起催化作用,而β和γ亚基在维持三聚体稳定性和作用底物特异性方面起重要作用。每个亚单位都存在由2-3种基因所编码的异构体(α1、α2、β1、β2和γ1、γ2、γ3),从理论上来讲,α、β和γ的不同异构体可形成各种可能的组合,可能有12种。α亚单位中的8个位点(苏氨酸172、苏氨酸258、丝氨酸485等)均可被磷酸化,其中苏氨酸172位点及其磷酸化对AMPK活性的调节起重要作用。α亚基的C-末端需要与β和γ-亚基结合。β亚基含有糖原中心结合域,C-末端需要与α和γ-亚基结合。包含可变的N-末端和四个保守胱硫醚β-内合酶的γ-亚基亚型与AMP和ADP结合。这些亚型在维持AMPK的稳定性和活性上发挥着重要的作用。AMPKα苏氨酸-172( Thr-172 ) 被磷酸化是AMPK激活所必需的。AMPK的γ-亚基暴露于α亚基激活位点(Thr-172) 可以引起构象变化。β亚基在失活或激活构象中充当结合α和γ-亚基的支架。研究表明,AMPK亚基不断地在神经元上表达,从而被功能活化。AMPK在细胞和全身水平调节着能量平衡。它开启以应对代谢压力,并被激素和细胞因子调控从而影响全身的能量平衡。一旦激活,AMPK在分解代谢途径中产生ATP。由AMP直接变构活化和上游激酶可逆性磷酸化AMPKα亚基的Thr-172均可调节AMPK的活性。AMPK磷酸化激活需要两种条件:γ-亚基暴露于α亚基激活位点(Thr-172)引起构象变化,α亚基的Thr-172被上游AMPK激酶在激活环路中磷酸化。主要机制包括LKB1复合体和CaMKKβ催化Thr-172磷酸化。相反,Thr-172可以被蛋白酸酶-2C去磷酸化失活。低血糖、缺氧缺血和热休克时细胞ATP减少,为增加AMP/ATP比,AMPK被激活。
作为低ATP水平的细胞能量感受器,AMPK调节着一些胞内系统:细胞糖摄入、脂肪酸的β氧化、脂肪酸和胆固醇的合成和葡萄糖转运酶4和线粒体生物合成等。通过抑制合成代谢能量消耗(脂肪酸和蛋白合成)和刺激能量产生,分解代谢(脂肪酸氧化和糖转运等)等途径,AMPK激活能增加细胞能量水平。然而,AMPK负调节着一些ATP消耗过程中的蛋白:调控CREB活性的转导蛋白-2( TORC2 )、糖原合成、固醇调节元件结合蛋白 (SREBP)、结节性硬化蛋白2(TSC2),抑制糖异生、糖原、血脂和脂质的合成。鉴于AMPK在脂质和代谢中的重要作用,其被作为治疗肥胖、糖尿病、心血管病、癌症、老化和神经退行性疾病的关键靶点。
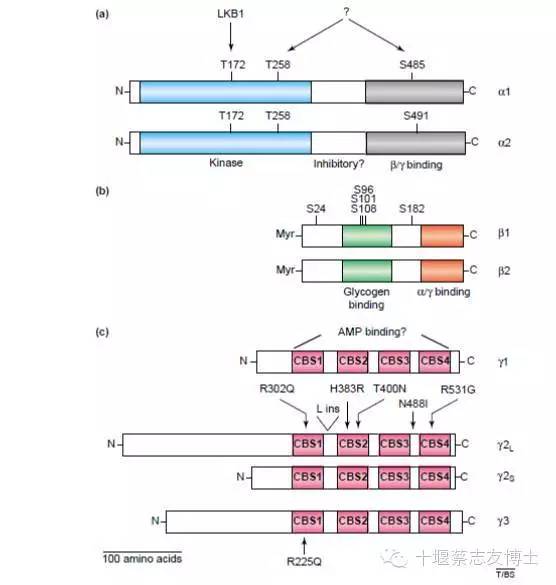 图1:AMPK结构
图1:AMPK结构
AMPK:AD脑能量代谢紊乱的标记物
流行病学、神经病理学和功能神经影像学证据显示了认知损害和AD患者的脑能量代谢紊乱。AD的神经退行性表现与脑能量代谢紊乱密切相关,包括胰岛素抵抗、糖摄取减少、线粒体功能障碍、胆固醇代谢障碍及Ca2+失衡等。高能量饮食和糖尿病对老化和AD的认知功能有害,而限制饮食能量对AD有益。研究显示AD患者皮质糖代谢明显降低,顶叶、颞叶和枕叶的皮质联合区受累明显。最新研究显示了AD的氧化应激增加、线粒体障碍、糖摄取受损。既往研究表明AD中氧化应激的增加可以损害神经元的能量代谢和离子平衡,对膜离子激动型ATP酶功能和糖以及谷氨酸转运蛋白造成损伤。这些氧化和代谢危害可以让神经出现兴奋性毒性和神经细胞凋亡的加速。
AMPK作为细胞和机体的代谢感受器可以感受ATP的水平。当ATP下降,AMPK被激活调剂细胞适应机体代谢变化。AMPK可以通过糖和脂质代谢刺激能量产生,相反也可以抑制蛋白和胆固醇合成等耗能功能。神经元AMPK信号激活主要靠胞浆AMP和Ca2+水平的增高。作为细胞能量代谢和感受器及调节器,AMPK在糖和脂质平衡中发挥着重要作用。AMPK活性随年龄增长而降低,导致老年相关的AD线粒体生物合成和功能下降。在AD的小鼠模型中,胰岛素样生长因子与AMPK间相互作用的紊乱可导致血管功能障碍。槲皮素可以通过激活AMPK来对抗高胆固醇饮食引起小鼠脑组织的氧化应激、炎症反应和Aβ沉积[3]。这些资料表明激活AMPK可以抑制氧化应激、改善AD线粒体功能和糖摄取,减少Aβ聚集,有利于减缓淀粉样斑块形成。
不少文献显示糖尿病与AD相关。越来越的证据显示胰岛素能提高AD患者认知,市面上的过氧化物酶体增殖剂激活受体γ(PPAR-γ)药物对AD早期患者有益[4]。通过AMPK依赖机制,罗格列酮减少了由NADH氧化酶介导的葡萄糖诱导的氧化应激。罗格列酮依靠PPAR-γ激活通过AMPK、Akt和JNK信号通路减少缺血损伤。在高脂饮食引起的血脂沉积和胰岛素抵抗老年大鼠,罗格列酮可以通过AMPK通路减轻胰岛素抵抗和增强脂质代谢。吡格列酮在体内可以刺激AMPK信号增加脂联素信号、线粒体功能和脂肪氧化。令人遗憾的是PPAR-γ激动剂因其严重心血管系统副作用而被退市。因此在包括AD在内的老年患者中使用该药物希望不大。当务之急是寻找临床前期的替代化合物。考虑到这类药物的风险,制药公司对此类研究的支持犹豫不前。过氧化物酶体增殖剂激活受体γ激动剂可以通过AMPK信号通路可改善AD患者认知损害和脑灌注。虽然其在糖尿病中高剂量使用,在AD患者长期治疗有效,但仍需阐明其长期应用的临床剂量和副作用。流行病学尚未完全揭示AD患者服用过氧化物酶体增殖剂激活受体γ激动剂的风险。在AD患者值得进一步研究低或高剂量长期治疗的效果。还有研究表明通过抑制AMPK,胰岛素抵抗可让星形胶质细胞能量供应不足和炎症加剧。因此,激活AMPK是改善脑能量代谢紊乱的一个潜在靶点。
二甲双胍是治疗糖尿病的一线药物,通常被认为是AMPK激活剂(可能通过LKB1)[4]。其很多作用的发挥也可能经由AMPK通路。有报道称二甲双胍诱发PP2A活性,减少体内体外依赖PP2A表型的Tau磷酸化。在胰岛素抵抗的体外模型“3型糖尿病”Neuro-2a神经元细胞系,长期高胰岛素状态可以出现AD标志性的病理改变,使用二甲双胍治疗可以阻止出现AD中分子和病理改变。这些资料表明二甲双胍在防止AD中具有潜在价值[5]。然而,也有发现二甲双胍激活AMPK可明显增加细胞内外Aβ片段,从而加剧AD[6]。尽管在人类身上尚未证实,二甲双胍在防治AD上还面临着许多挑战,AMPK在AD中的作用需要进一步研究。
AMPK调节Aβ病理生理过程
AD已被确定为蛋白质错误折叠疾病,引起的异常折叠Aβ蛋白在大脑沉积。Aβ有39-43个氨基酸是一种淀粉样蛋白前体,是能穿透神经细胞膜的跨膜蛋白。Aβ的生成要求两序列裂解反应,两种蛋白水解酶:β分泌酶和γ分泌酶。在淀粉样降解途径,Aβ产生始于β分泌酶介导的APP残端的Met671与Asp672裂解,裂解成一个可溶性细胞外片段(sAPPb)和细胞的膜结合片段(C99)。接着C99由BACE通过γ分泌酶在疏水性跨膜区域Val711或Ile713进一步裂解,产生Aβ。在非淀粉样降解途径,APP经α分泌酶产生sAPPα阻止Aβ生成。相反γ分泌酶裂解后β分泌酶再裂解也可产生Aβ。有关β/γ分泌酶裂解抑制剂的研究表明其可能成为一个抗AD的有效方法。
许多资料表明AMPK是Aβ生成的关键调节剂。不少研究表明AMPK在Aβ生成的的病理过程中发挥着重要作用。最近研究表明在AMPKα2敲除的神经元Aβ生成增加。AMPK激活可能会通过调节APP生成而减少Aβ生成(图2)[7]。
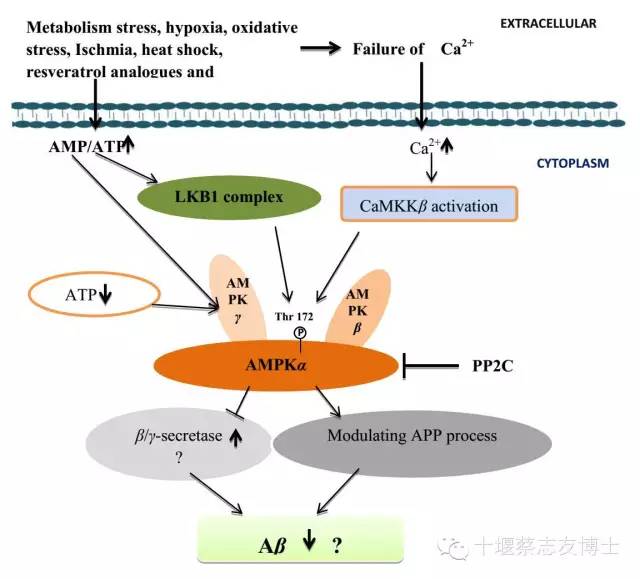 图2:AMPK调节Aβ生成的可能作用机制。
图2:AMPK调节Aβ生成的可能作用机制。
AMPK参与胆固醇平衡仅次于HMG-CoA还原酶,调节胆固醇和鞘脂的代谢。胆固醇和鞘脂与调节APP加工和Aβ产生有关,从而参与到AD的病理过程。Won等在AMPKα2敲除的小鼠神经元上观察到高水平的胆固醇和鞘脂,AMPK调节胆固醇和鞘脂的水平从而调剂APP分别,进而其代谢引起Aβ的产生[8]。因此,AMPK通过调节胆固醇和鞘脂的水平就APP分布从而导致Aβ的产生。
AD患者的瘦素是下降的。Greco等报道显示瘦素治疗的人群和大鼠神经元细胞通过激活AMPK抑制Aβ的产生[9]。直接用细胞渗透性活化剂5-氨基咪唑-4-甲酰核苷酸(AICAR)刺激AMPK可复制瘦素的作用,降低Aβ产生,而AMPK的抑制剂CompoundC可抑制瘦素活性增加Aβ生成。最近的研究证明,一种天然多酚白藜芦醇通过钙/CaMKKb,提高AMPK活性从而在体内和体外能激活几种代谢传感器和降低Aβ的水平。AMPK抑制剂则可以降低白藜芦醇对Aβ水平的影响。白藜芦醇可以通过作用于mTOR抑制AMPK从而触发自噬溶酶体降解Aβ[10]。Kwon等发现褪黑素通过调节AMPK通路能增强白藜芦醇的抗Aβ诱导的神经退行性病变效果[11]。大量体内外模型显示褪黑素不仅能抑制Aβ的产生,还可以阻止结构依赖性相互作用淀粉样蛋白纤维形成。Kwon等研究还表明褪黑素和白藜芦醇能显著降低ROS产生和线粒体膜电位破坏,减轻Aβ42诱导的神经毒性。Aβ42处理能激活AMPK活性,而褪黑素和白藜芦醇治疗能抑制的AMPL磷酸化[12]。Aβ42可显著增加AMPKα1活性而对AMPKα没有明显影响。Thornton等研究显示CaMKKβ需要Aβ42激活AMPK。研究显示Aβ42先通过CaMKKβ信号通路调节AMPK,接着激活AMPKa1复合物[13]。总而言之,这些资料显示CaMKKβ介导的AMPK激活能阻止Aβ进一步生成,可能在AD病理中发挥抗Aβ毒性作用。
已被证明的是AMPK的活化是通过抑制BACE1表达从而调节APPβ-裂解进而减少Aβ的生成。Lu等发现AMPK激活能显著降低高胆固醇喂养老年大鼠胆固醇水平,下调BACE1表达,降低脑组织Aβ沉积,腹腔注射AMPK抑制剂CompoundC能抵消这种作用[14]。
排除血糖和胰岛素因素,AMPK激活能上调BACE1表达从而由淀粉样前体蛋白产生Aβ。Chen等研究显示二甲双胍通过上调BACE1的转录激活AMPK,能显著增加细胞内外Aβ的产生。AMPK抑制剂CompoundC能显著抑制BACE1转录和Aβ生成揭示了这个AMPK依赖机制[15]。此外,越来越多数据表明5-氨基咪唑-4-甲酰核苷酸(AICAR)介导AMPK激活,通过抗炎和抗氧化的调控降低Aβ。最新研究表明AICAR治疗可降低Aβ的产生,这可能是通过降低脂质APP水平而不是调节α或β分泌酶活性发挥作用的。Won等观察AICAR治疗下调神经元C99水平,没有改变体外APP或BACE1水平及α或β分泌酶活性。这表明AICAR介导的APPβ裂解减少与细胞APP和BACE1水平及α或β分泌酶活性相互独立[16]。
总之,AMPK激活到底是抑制BACE1表达调节APPβ裂解减少Aβ产生,还是上调BACE1增加Aβ产生存在争议。AMPK激活与Aβ产生是敌是友,较大程度取决于实验条件和生化设置。AMPK是否是对抗Aβ产生的强有力靶点值得进一步研究。
AMPK调控tau蛋白磷酸化
Tau高度磷酸化的Tau蛋白假说被认为是AD的主要病因,Tau蛋白沉积为配对的螺旋丝,交互聚集成神经原纤维缠结和有毒的可溶性Tau蛋白片段。脑脊液中升高的磷酸化tau是AD相关脑组织病变的一个可靠生物标志物。脑脊液Aβ42水平异常降低与高脑脊液tau蛋白水平的组合对诊断AD颇有帮助。在一般情况下,tau蛋白在丝氨酸和苏氨酸残基超磷酸。Tau蛋白磷酸化由一系列激酶调节,包括糖原合成酶激酶3、细胞分裂蛋白激酶5、SRC家族酪氨酸激酶、蛋白激酶A、CaMKKβ,应激活化蛋白激酶、微管蛋白激活的蛋白激酶、丝裂原活化蛋白激酶以及AMPK。研究表明AICAR直接刺激AMPK抑制tau蛋白磷酸化,相反AMPK抑制剂compoundC,可以增加tau蛋白磷酸化。这些表明是AMPK是tau蛋白磷酸化的一个关键的调节因素。
AMPK是一种生理tau蛋白激酶,磷酸化多个tau蛋白抗原表位。Thornton等发现在原代小鼠皮层神经元,AMPK增加tau蛋白Ser262磷酸化,影响tau蛋白微管结合能力[17]。为弄清Ser262是否是AMPK对tau蛋白主要的磷酸化位点,Thornton等建立了His标记WT野生型tau蛋白或构建携带Ser262至S262A突变的CL13细胞系。Thornton发现62a突变对GSK-β(一个tau激酶)磷酸化tau蛋白的能力无明显影响。与WT野生型相比,AMPK引起的tau蛋白磷酸化存在于s262a突变。这表明AMPK在Tau蛋白有额外的AMPK磷酸化位点。Thornton还发现AMPK通过改变tau蛋白微管结合在Ser396和Ser262位点磷酸化tau蛋白[17]。通过CaMKKβ介导活化途径AMPK上游的NMDA受体,AMPK在tau蛋白磷酸化的病理生理发挥着重要作用。
最近的研究发现,AMPK在体外可以直接在thr-231和ser-396/404磷酸化tau蛋白。Vingtdeux等发现在3R(3重Tau蛋白)和4R(4重Tau蛋白)tau蛋白病(包括AD)脑神经元有激活的AMPK(p-AMPK)异常积累[18]。在AD脑组织中,p-AMPK在神经纤维沉积,淀粉样蛋白斑块周围的轴突营养不良,90%以上神经元存在纤维缠结或纤维缠结前体。在纯双螺旋结构不存在p-AMPK,这表明p-AMPK并不与缠结的Tau蛋白共聚合。这些数据表明AMPK激活之前tau蛋白沉积。
AMPK/mTOR/自噬/AD
自噬是一种进化上保守的溶酶体依赖通路和细胞内自我保护过程,降解长寿蛋白或错误折叠的蛋白质和受损的细胞器。很显然,在健康的神经元细胞自噬积极和高效,在AD中自噬最有可能来自受损的自噬泡间隙而不仅仅是诱导强自噬。一种慢性退化的神经元自噬溶酶体系统可能是从正常脑老化到病理性老化再到阿尔茨海默氏病神经退行性疾病过渡的一个关键因素。在阿尔茨海默氏症的蛋白质清除不当可能导致的自噬溶酶体降解途径的损坏,或诱导这个通路的改变,这可能导致神经功能障碍和神经元丢失。事实上,自噬是参与清除Aβ和tau蛋白异常聚集的一个关键途径。诱导自噬在体内体外能增强可溶性和聚合形式Aβ和tau蛋白的清除。因此,控制自噬溶酶体蛋白降解是AD的一个潜在治疗靶点[19, 20]。
mTOR是一个289kDa的丝氨酸/苏氨酸蛋白激酶的PIKK家族的成员,调节细胞生长,细胞增殖,细胞运动,细胞的存活率,蛋白质的合成,转录,是一个广泛的细胞信号。mTOR与其他蛋白质相互作用,形成复杂的两种主要类型,mTOR复合物1和2(mTORC1和mTORC2)。越来越多的证据表明,自噬是通过mTOR负调控,自噬是受mTORC1调控第二个关键过程。营养丰富的条件下,mTOR被召集至溶酶体表面,火化后就可以促进细胞生长,抑制细胞自噬。数据还表明,mTOR作为中央控制器在AD的发病机制中起着重要作用,降低mTOR通路可诱导Aβ产生和积累的tau蛋白。mTOR上游和下游信号通路成分参与到各种各样的AD发病机制。mTOR抑制剂雷帕霉素能改善AD小鼠模型学习记忆障碍,减轻Aβ和tau蛋白的病理改变。mTOR信号通路是通过自噬途径调节Aβ和tau蛋白一个关键调节因素。
众所周知,AMPK通过直接磷酸化ULK1促进自噬。而mTOR这个结合生长因子和营养信号中央细胞生长调节因子,能抑自噬。新近的研究表明,AMPK通路能调节mTOR的活性。最近的报告表明,mTOR通通过CaMKKβ介导激活AMPK可增加胞内钙,从而诱导自噬。
最近的研究表明,AMPK激活降低mTOR信号通路活化有利于细胞自噬,从而促进溶酶体降解Aβ(图3)[21, 22]。Vingtdeux等研究显示,白藜芦醇通过增加细胞内钙水平激活AMPK,通过CaMKKβ促进thr-172AMPK磷酸化,从而通过抑制mTOR诱导自噬降低Aβ水平,在体外通过溶酶体系统促进胞内Aβ降解[23]。Vingtdeux等还发现RSVA314和RSVA405两种白藜芦醇类似物,发挥着白藜芦醇同样的作用机制:促进CaMKKβ依赖性激活AMPK,抑制mTOR,促进自噬和溶酶体降解Aβ[23]。
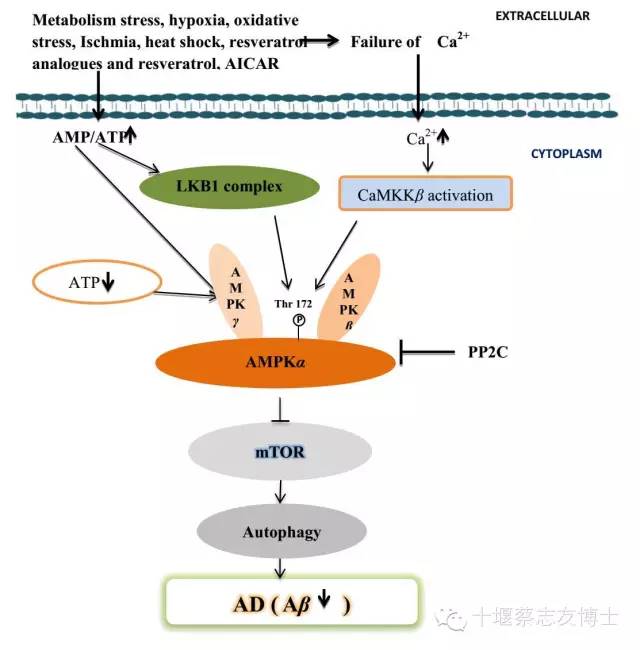
图3:通过mTOR和自噬途径AMPK调节Aβ生成的作用机制。
结论与展望
AMPK这个脂质和糖代谢的重要调节因子,被认为是代谢相关疾病如AD的一个重要治疗靶点。如前所述,脑的能量代谢紊乱参与了AD的发病机制。AMPK活化能增强紊乱的脑能量代谢,参与了AD的发病机制。AMPK的激活还可以通过调节淀粉样蛋白前体产生减少Aβ生成。由于AMPK是一种生理tau蛋白激酶和tau蛋白磷酸化表位多,通过AICAR直接刺激AMPK可抑制tau蛋白磷酸化。AMPK的激活可抑制mTOR信号通路,从而促进自噬,进而溶酶体降解Aβ。研究表明,AMPK是抗代谢应激的神经保护因子,对AD的预防作用显著。AMPK在AD的发病机制中起着重要的作用,是AD的一个潜在治疗靶点。
然而,研究还表明,AMPK的活化具有神经保护作用,还可能导致的不利结果。有研究表明,AMPK激活可能是有害的,给予抑制AMPK的药物对中风有神经保护作用。AMPK激活可能是通过加剧缺血诱导的代谢衰竭对实验性脑卒中有害。研究证据也显示,AMPKα-2参与了脑卒中AMPK活化所致的有害影响。亨廷顿氏这种病纹状体神经退行性疾病AMPKα-1过度激活。在AD和其他tau蛋白病的神经元,AMPK是异常激活可直接在Thr-231和ser-396/404磷酸化纤维缠结或纤维缠结前体的tau蛋白。因此,AMPK与Aβ的产生和tau蛋白聚集存在着亦敌亦友的关系。考虑到AMPK的保护性能,AMPK激活是否导致细胞存活或消亡的活动在很大程度上取决于条件和生化的设置。它可以由实验设置有效的抑制剂和AMPK激活剂从而产生有争议的结果。对药理操作后得到的AMPK结果的解释,AMPK实际发挥的作用更重要的是应考虑到实验条件而非应用活性化合物。因为一般的AMPK激活剂或抑制剂的作用取决于实验的设置,而不能一概而论。不管怎样,将来的研究需要放在AMPK在AD发病或AD治疗中的作用上。
AMPK活化是试图恢复细胞稳态的一种保护性反应。AMPK通路的激活可能是治疗和预防各种疾病的关键,然而AMPK过度激活可能是有害的。AMPK通路是通过调控自噬途径调节Aβ和tau蛋白一个关键因子。先前在阿尔茨海默病中AMPK相关物质的发现可能有深远的临床意义。不过,目前仍不清楚AMPK是否发挥致病作用,保护作用,或者是阿尔茨海默病的一个过程。此外,激活AMPK对人类临床是否有益仍是一个待解的问题。因此,有必要对AMPK生物化学和生理学进行进一步揭示,对于AD防治的AMPK相关药物机制有一个更好的了解。
本文转载自梅斯医学,如有侵权,请联系我们删除!